Atherogenic Dyslipidemia
Defining CMR - Visceral Adipose Tissue: the Culprit? Complications of Visceral ObesityKey Points
- Chylomicrons (dietary) and VLDL (endogenous) are the main triglyceride carriers in the blood. LDL and HDL are the main cholesterol carriers.
- LDL delivers cholesterol to peripheral tissues. HDL delivers cholesterol from tissues to the liver.
- The “traditional” dyslipidemia typically associated with visceral obesity includes hypertriglyceridemia, the presence of small, dense LDL, and low HDL cholesterol levels. All three abnormalities are metabolically linked.
- Chylomicron and VLDL remnants (not routinely measured) are also elevated in visceral obesity.
- Under pro-atherogenic conditions, chylomicron remnants, VLDL remnants, and small LDL deliver cholesterol to the artery wall (pro-atherogenic). HDL removes cholesterol from the artery wall (anti-atherogenic).
- Insulin resistance plays a key role in linking visceral obesity and dyslipidemia.
- A newer set of metabolic risk factors—the “atherogenic metabolic triad”—includes hyperinsulinemia, hyperapo B, and small LDL particles.
- These risk factors enable additional subjects at risk of CVD to be identified.
- A number of drugs are available to treat each component of the dyslipidemia of visceral obesity. But a key therapeutic objective remains the reduction of visceral fat, the primary cause of dyslipidemia.
Overview
Atherogenic dyslipidemia is one of the metabolic abnormalities that define the metabolic syndrome, the cluster of cardiovascular risk factors frequently associated with visceral obesity. This section outlines the components of the dyslipidemia that characterizes visceral obesity and the metabolic syndrome, as well as their causes and impact on cardiovascular disease (CVD) risk. A brief review of the intravascular lipid transport system will help place the dyslipidemia of visceral obesity in proper context.
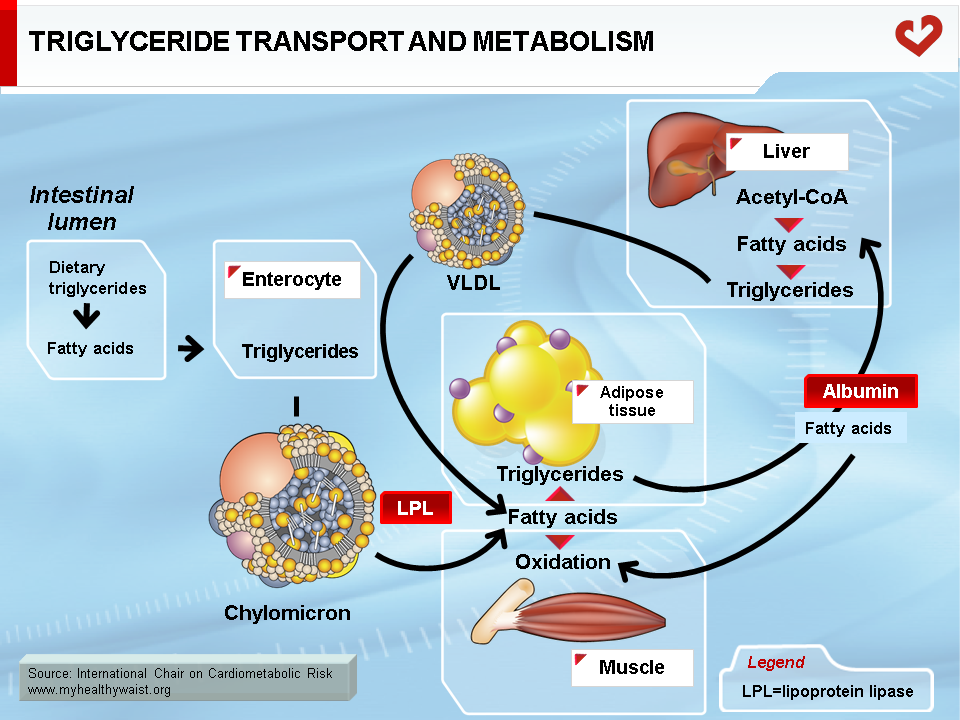
The major lipids involved in dyslipidemia are cholesterol and triglycerides. These lipids are insoluble in blood (like oil in water) and must be packaged into macromolecular lipid-protein complexes called lipoproteins for export to other organs (Figure 1). Lipoproteins are classified according to their lipid and protein composition. The major lipoprotein classes are as follows: chylomicrons, very-low-density lipoproteins (VLDL), remnants (or intermediate-density lipoproteins, IDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL). Chylomicrons and VLDL are the main transporters of dietary and endogenous triglycerides respectively, while LDL and HDL mainly carry cholesterol between the liver and extrahepatic tissues [1-4]. Following is a brief, simplified overview of the metabolic fate of each of these lipoprotein families (Figures 2 & 3).
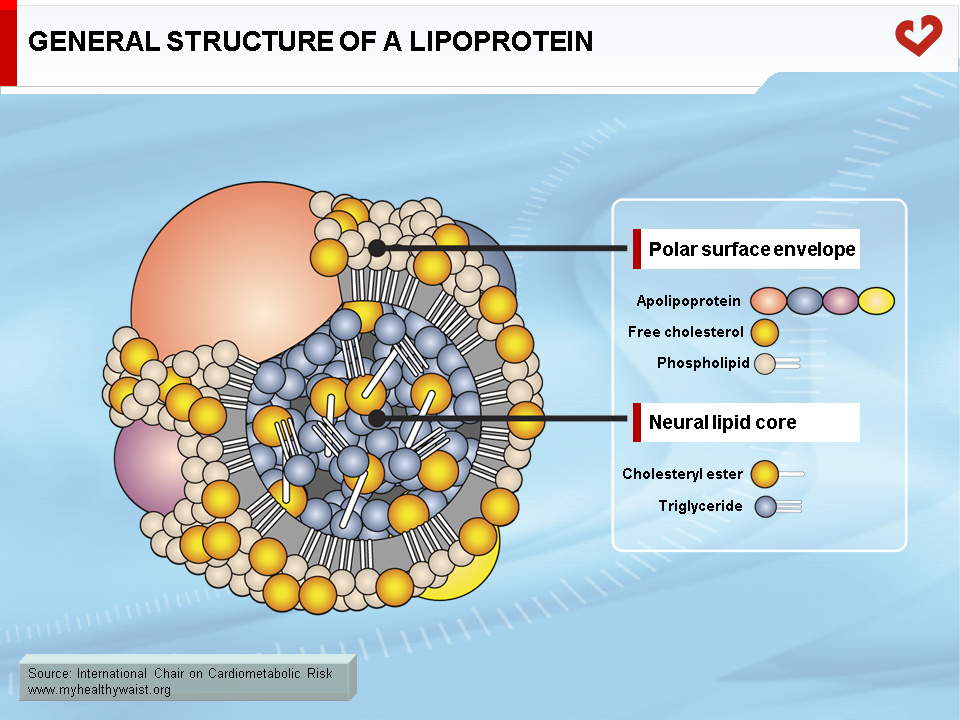

Intravascular Lipid Transport in the Fasted State
The liver produces endogenous lipids and provides peripheral tissues with cholesterol for membrane assembly and steroid hormone synthesis and triglycerides for storage (adipose tissue) or as a source of energy in oxidative tissues (skeletal muscle, heart). The liver is also where cholesterol is excreted from the body.
VLDL Metabolism
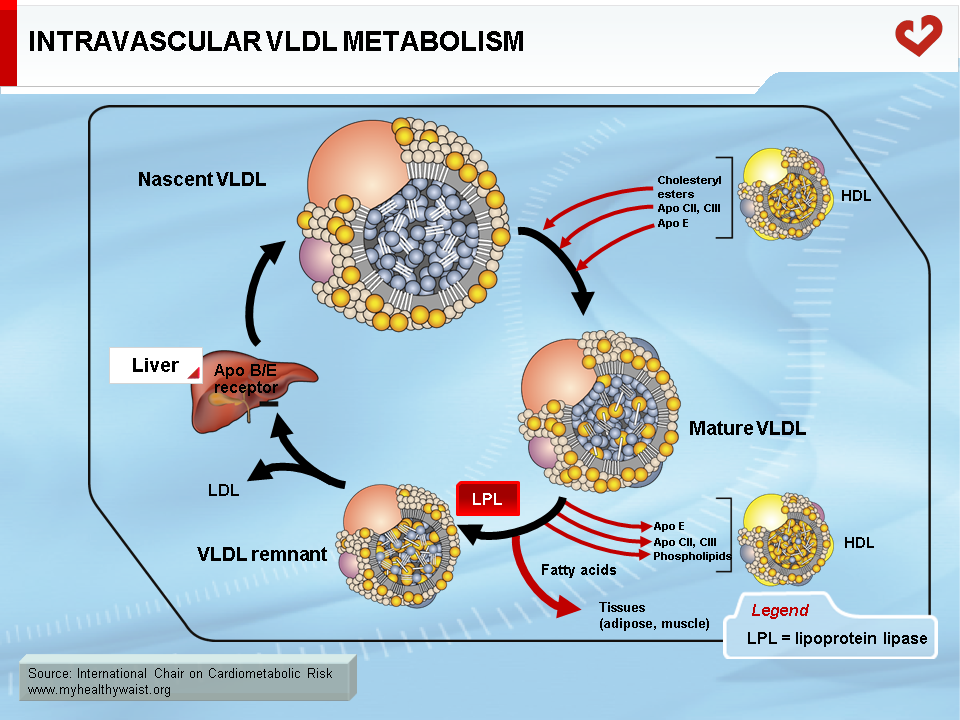
Endogenous cholesterol and triglycerides are produced in the liver and exported to the blood within VLDL. The major role of VLDL is to deliver cholesterol and triglycerides to extrahepatic tissues (after VLDL becomes a LDL, as described below). Liver VLDL assembly is a complex process whereby apolipoprotein (apo) B100 gradually acquires lipids through the action of microsomal triglyceride transfer protein (MTP) and ADP ribosylation factor 1 (ARF-1) [5]. The number of VLDL particles synthesized and their composition depend on two main factors: lipid availability inside the hepatocyte and hormonal control, mainly by insulin. Apo B is expressed constitutively: when hepatic lipid stores are low, apo B is less lipidated and undergoes degradation within the hepatocyte. Apo B100 is the major apolipoprotein of VLDL and LDL and is essential for hepatic lipoprotein synthesis. There is only one apo B per lipoprotein, and plasma apo B concentration is a measure of particle number, a variable that may be quantified for clinical purposes. In the liver, lipid abundance (such as in visceral obesity and hepatic steatosis) promotes apo B lipidation and VLDL formation. Nascent VLDL is secreted into the bloodstream in two forms: VLDL1 (larger, with more triglycerides) and VLDL2. In the bloodstream, they acquire other apolipoproteins to become mature particles. Both forms of VLDL undergo extensive remodelling within the bloodstream (Figure 4). During their journey, VLDL particles encounter lipoprotein lipase (LPL) [6]. Bound to the endothelial cells of blood capillaries, this enzyme hydrolyzes triglycerides carried by VLDL, gradually reducing its size until it becomes a triglyceride-depleted VLDL remnant. Fatty acids generated by LPL from VLDL triglycerides are absorbed by tissue cells in contact with the capillaries. These acids are either stored as triglycerides (mainly adipose tissue) or used as an energy source (in oxidative tissues such as skeletal muscle and the heart).
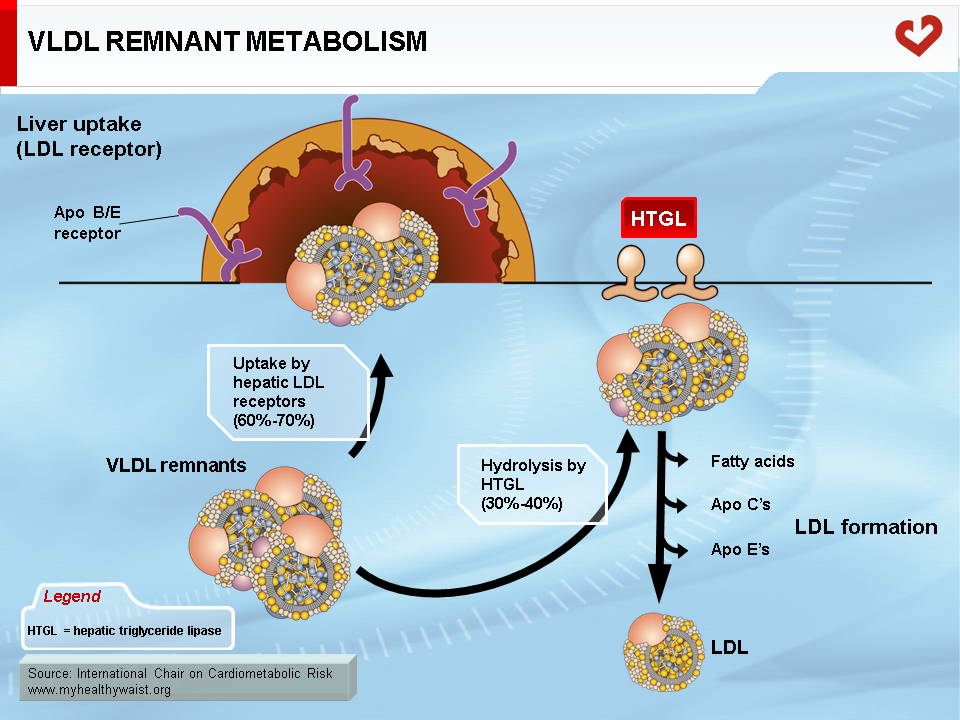
The VLDL Remnant-LDL Cascade
The VLDL remnant (also called IDL) follows one of two metabolic routes (Figure 5). Many VLDL remnants bind to LDL receptors (see below) at the surface of liver cells, are internalized, and then broken down into their various components. The remnants not taken up by the liver interact with hepatic triglyceride lipase (HTGL), which is found inside liver capillaries and further hydrolyzes most of the remaining triglycerides (and some phospholipids). This creates a cholesterol-rich, triglyceride-depleted particle called LDL [1].
LDL Metabolism
LDL is the major cholesterol carrier in human blood. Although the vast majority of tissues make their own cholesterol, the liver provides some cholesterol through the VLDL remnant-LDL cascade (Figure 5). Circulating LDL binds to the LDL receptor (LDLr) found in many tissues [7]. The entire LDL is then internalized and degraded, providing the cell with cholesterol. Many LDL particles bind to liver LDLr, thereby closing a futile cholesterol cycle (Liver → VLDL → LDL → Liver). The remaining particles are taken up by various tissues where their cholesterol component becomes part of cell and organelle membranes or serves as a precursor in endocrine glands that synthesize steroid hormones such as cortisol, estrogen, and testosterone.
High LDL cholesterol in the blood is a strong CVD risk factor because LDL can enter the arterial wall and contribute to the formation of atherosclerotic plaque. High LDL cholesterol is not, however, a dyslipidemia characteristic of visceral obesity and the metabolic syndrome.
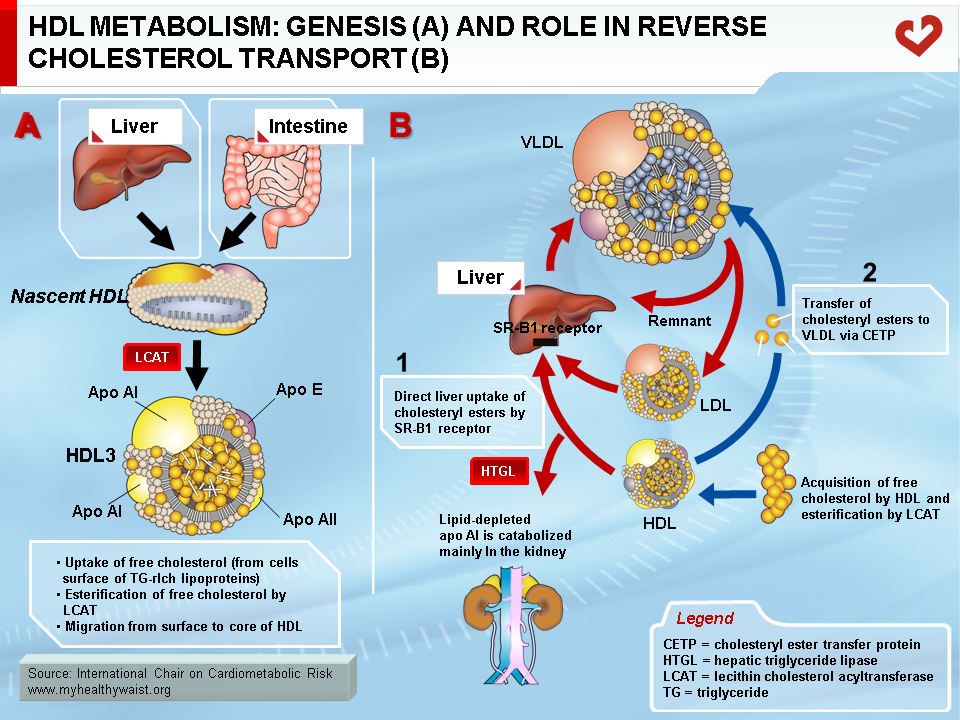
HDL Metabolism
With the exception of the liver, all tissues are unable to degrade and dispose of cholesterol as byproducts. Surplus cholesterol (e.g., from dead cells or local production) must therefore be excreted from cells and transported to the liver for disposal in the bile. This is carried out by HDL in a process called reverse cholesterol transport [1]. HDL is secreted into the bloodstream in nascent, discoidal form and is mainly composed of apo AI or apo AI and AII and phospholipids (Figure 6A). Cellular cholesterol to be disposed of is presented to HDL at the cell surface by sterol receptors, with ABCA1 playing a key role. ABCA1 transfers free (unesterified) cholesterol to the surface of the HDL particle, where the enzyme lecithin cholesterol acyltransferase (LCAT) adds a fatty acid molecule to the cholesterol, which then becomes esterified. This is a very insoluble form of cholesterol and it moves to the core of HDL, making room at the surface for more free cholesterol uptake from cells. HDL thus becomes gradually enriched in cholesterol and takes a spherical shape. HDL delivers some of its core cholesterol esters to the liver by interacting with another receptor, SRB1. The smaller HDL returns to the circulation in a new cycle of cholesterol uptake and delivery. The particle will eventually be cleared from the bloodstream by the liver or kidneys (Figure 6B). The smaller the HDL particle, the shorter its time in the bloodstream and its contribution to reverse cholesterol transport. Once in the liver cell, cholesterol can follow one of several paths: storage in vesicles, re-secretion within VLDL, or excretion in the bile in its free form or as bile salts, which help fat emulsion and absorption in the gut. Some of the cholesterol now in the gut is reabsorbed and returns to the liver, while the remaining cholesterol is excreted from the body.
High blood levels of HDL cholesterol help protect against atherosclerosis. Among other functions, HDL removes cholesterol from the arterial wall, counteracting LDL deposition and reducing plaque formation.
Further Intravascular Remodeling of Lipoproteins
As discussed above, all lipoproteins undergo extensive remodelling in the bloodstream by the enzymes LPL, HTGL, and LCAT. Other enzymes remodel the lipoproteins further. Endothelial lipase (EL), the physiological role of which remains ill-defined, appears to act mainly on HDL to remove some phospholipids and modify HDL composition [8]. Other phospholipases (e.g., sPLA2-IIA) also remodel some lipoproteins in a similar way. The contribution of EL and other phospholipases to cardiovascular risk remains uncertain. Studies have suggested that higher levels of EL and sPLA2 can increase CVD risk through problematic HDL remodelling. Another intravascular enzyme, cholesterol ester transfer protein (CETP), also remodels lipoproteins [9]. CETP facilitates the exchange of lipids between lipoproteins in the following manner: triglyceride-rich lipoproteins (VLDL) transfer triglycerides to cholesterol-rich lipoproteins (LDL, HDL) in a reciprocal exchange for cholesterol. Triglyceride-rich lipoproteins thus gain some cholesterol while cholesterol-rich lipoproteins gain triglycerides. This remodelling affects lipoprotein metabolism, as discussed below.
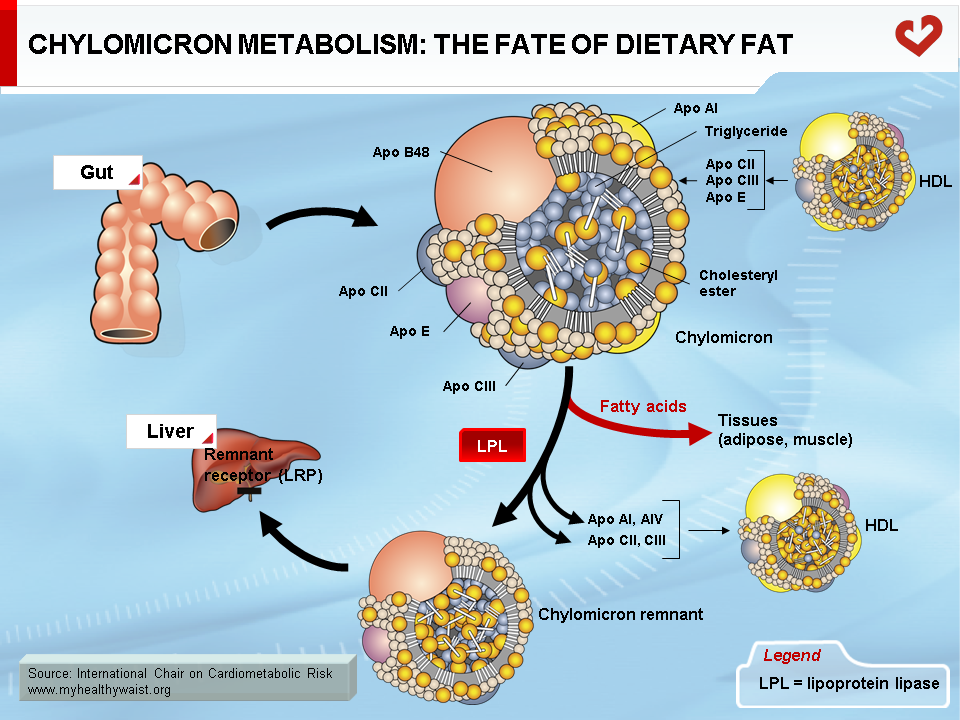
Intravascular Transport of Dietary Fat
Dietary fat, which is mainly composed of phospholipids (not addressed here), triglycerides, and cholesterol, is taken up by the intestinal cells. Though the gut is very efficient at absorbing triglycerides (close to 100%), it is less efficient at absorbing cholesterol and absorption can range from 1/3 to 2/3 of the amount ingested. Some drugs interfere with intestinal cholesterol absorption and play a role in lowering blood cholesterol levels. Once absorbed by the intestinal cells, dietary lipids are packaged into lipid-rich lipoproteins known as chylomicrons through a process resembling that of VLDL synthesis in the liver [10]. Chylomicrons differ from VLDL in that their major apolipoprotein is apo B48 instead of B100 and their lipid content is much greater than the liver-derived particles. Once in the bloodstream, chylomicrons are acted upon by the enzyme LPL (exactly as VLDL), which hydrolyzes the triglyceride moiety of chylomicrons to produce fatty acids that are then taken up by the various tissues containing LPL (Figure 7). The resulting particle, the chylomicron remnant, is thus depleted of triglycerides and relatively richer in cholesterol. It has become small enough to leave the blood capillaries and come into contact with a receptor, LDL-related protein (LRP), located on the surface of liver cells. The chylomicron remnant is then internalized and degraded, delivering to the liver its load of dietary cholesterol.
The basic processes of fasting lipoprotein metabolism continue in the postprandial state. However, in healthy people, several adaptations take place upon food ingestion, mainly to reduce the output of endogenous lipids into the circulation and route dietary lipids to the appropriate tissues. Insulin plays a key role in these adaptations. As seen above, insulin tends to reduce VLDL secretion. This is amplified by the postprandial rise in insulin. After a meal, the liver therefore puts out fewer triglycerides, making LPL more available for hydrolysis of chylomicron-bound dietary triglycerides. The postprandial rise in insulin also strongly amplifies the hormone’s lipolysis-inhibiting action in adipose tissue, which greatly reduces the release of fatty acids into the bloodstream. In addition, higher insulin levels switch LPL activity, which increases in adipose tissue at the expense of muscle tissue to favour storage of the incoming chylomicron-bound dietary fat [6,11-13].
The Dyslipidemic Profile of Visceral Obesity and the Metabolic Syndrome
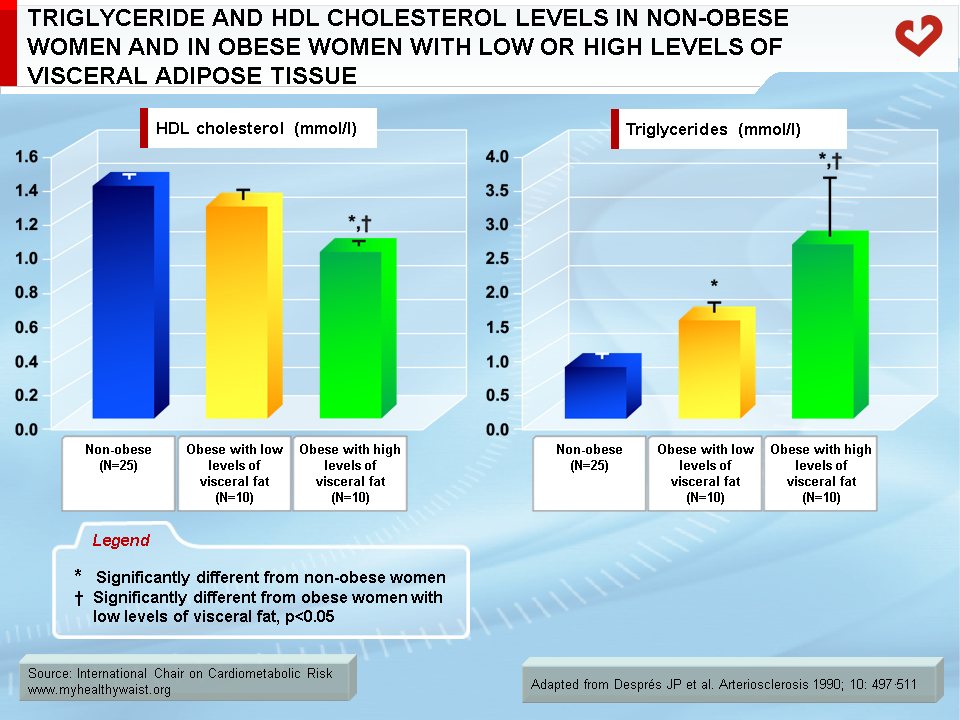
Although overweight and obesity are linked to abnormalities in lipoprotein metabolism, it is now well established that it is excess visceral fat that has the strongest ties to these abnormalities [14-20]. It has been known for several decades that obesity is frequently associated with a dyslipidemic state that includes increased triglyceride concentrations and reduced HDL cholesterol levels. However, because obesity is remarkably heterogeneous, not every obese patient is dyslipidemic while some moderately overweight individuals clearly are. Studies conducted over the last two decades have shown that in both men and women, obese patients matched for total adiposity but with either low or large amounts of visceral adipose tissue were markedly different in their fasting lipoprotein-lipid profile [21,22]. For instance, obese patients with low levels of visceral adipose tissue had a normal lipoprotein-lipid profile compared to lean controls, while equally obese patients with high levels of visceral fat had the high triglyceride, high apolipoprotein B, low HDL cholesterol, and small, dense LDL and HDL atherogenic dyslipidemic state (Figure 8). This dyslipidemic state of visceral obesity is a key feature of insulin resistance and the clustering abnormalities of the metabolic syndrome.
Main Features of the Obesity-related Dyslipidemic Profile: Fasting State
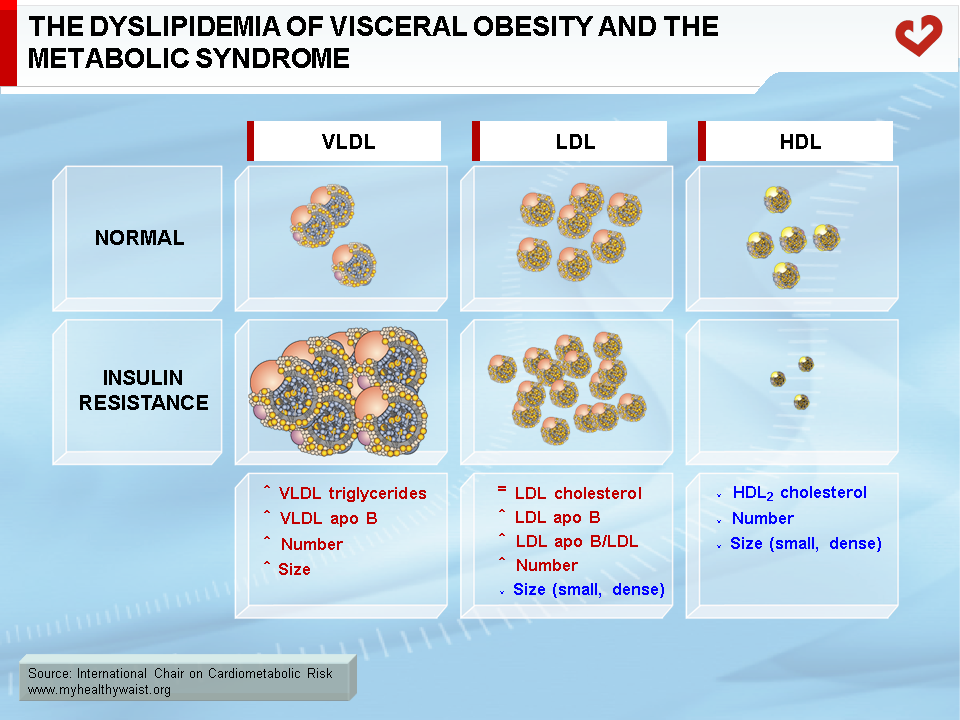
The dyslipidemia that partly defines the metabolic syndrome includes the following abnormalities: high levels of triglycerides, low levels of HDL cholesterol, and relatively normal total and LDL cholesterol levels but more LDL particles (as quantified by high apo B levels) that are smaller and denser than normal (Figure 9). In abdominal obesity, HDL particles are also small in size because of the presence of hypertriglyceridemia [23]. Total and LDL cholesterol levels are generally within the normal range unless unrelated abnormalities are present [19]. In a typical clinical setting, hypertriglyceridemia and low HDL cholesterol will therefore be the two major detectable features of the dyslipidemia associated with visceral obesity. The combination of high triglycerides, small and dense LDL, and low HDL cholesterol is known as the “lipid triad” and recognized as a major CVD risk factor. The various components of the lipid triad are closely linked to each other, as outlined below. The lipid triad and LDL cholesterol are considered to be more traditional CVD risk factors. With visceral obesity, alternative methods of grouping risk factors provide a surer way to identify people at risk than traditional lipid risk factors. Such is the case for the “atherogenic metabolic triad,” which includes hyperinsulinemia, hyperapo B, and small LDL particles [24]. The present document focuses on the lipid abnormalities associated with visceral obesity.
Visceral obesity and the metabolic syndrome are also characterized by high fasting blood levels of “non-lipoprotein” lipids, namely nonesterified (or free) fatty acids. Blood fatty acid levels are largely determined by their rate of output by adipose tissue. This explains why excess fat, especially in the highly lipolytic visceral depots, maintains high levels of nonesterified fatty acid levels in the portal vein draining into the liver. Visceral fat therefore increases delivery of fatty acids to the liver. Subcutaneous fat in the abdominal region is thought to be the main source of increased systemic (general circulation) fatty acid levels. Fatty acids are readily taken up by tissues. When levels are too high inside cells, fatty acid derivatives are toxic to tissues such as muscle, liver, heart, and pancreas and may seriously harm their function. This “non-lipoprotein” dyslipidemia therefore contributes to lipotoxicity and plays an important role in the development and maintenance of insulin resistance [25].
Causes of Hypertriglyceridemia
Hypertriglyceridemia is central to the dyslipidemic profile of visceral obesity. It is caused by a combination of increased liver VLDL triglyceride production and impaired clearance from the circulation. As discussed above in the section pertaining to VLDL production, fatty acid availability within the hepatocyte and insulin are two major modulators of VLDL assembly and secretion. In people who are viscerally obese, their expanded visceral fat depots produce more fatty acids (through lipolysis) that drain into the portal vein. The liver is thus exposed to large amounts of fatty acids, many of which are readily taken up by liver cells. These fatty acids ultimately contribute to the synthesis of triglycerides, which are then incorporated into VLDL particles and secreted into the circulation, raising plasma triglyceride and apo B (more VLDL particles) levels. It is the large VLDL-1 subpopulation that is most affected. Therefore, fat accumulation in the liver (steatosis) due in part to increased adipose-derived fatty acids is a major contributor to the hypertriglyceridemic state of visceral obesity [11-13].
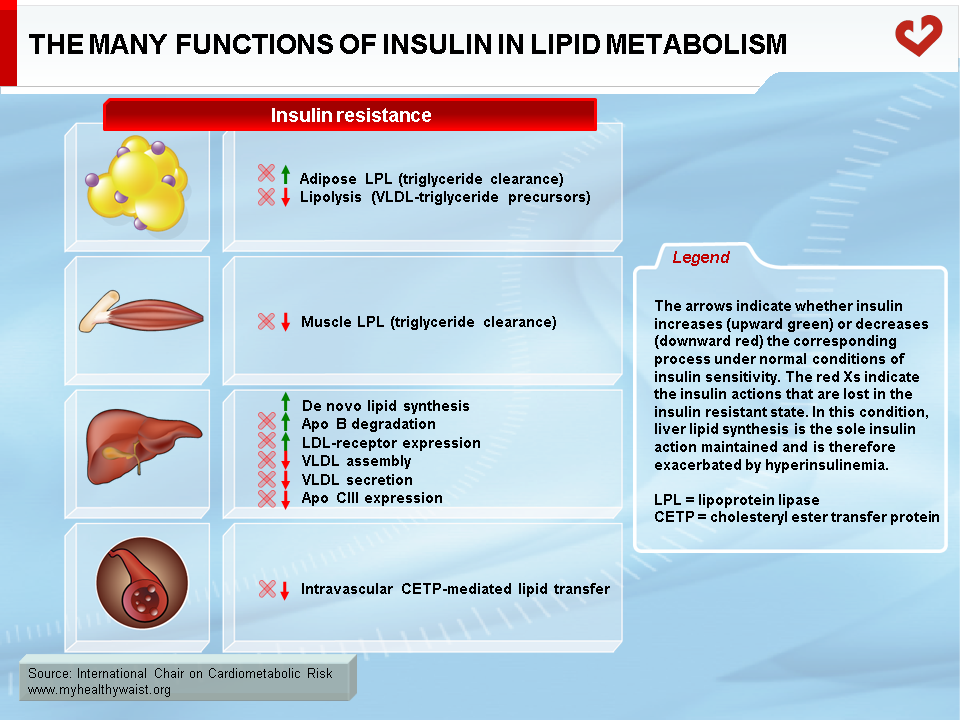
Abnormal insulin action in the liver is another key factor underpinning excessive VLDL production and attendant hypertriglyceridemia. In healthy persons, insulin tends to promote the production of new fatty acid molecules in the liver. This effect, however, is quite modest. In viscerally obese, insulin-resistant persons, higher amounts of circulating insulin accelerate lipid synthesis in the liver. This causes steatosis and raises VLDL secretion along with lipids originating from visceral fat. In addition, despite its ability to promote lipid synthesis, insulin normally tends to reduce hepatic VLDL output into the circulation. This effect is lost in insulin-resistant persons, which removes a barrier to VLDL secretion and thereby enables it to increase [11-13].
An additional contributor to hypertriglyceridemia is impaired clearance of VLDL and its remnants from the circulation. Insulin resistance reduces the global availability of the triglyceride-hydrolyzing enzyme LPL. Combined with subtle changes in VLDL composition, this increases the residence time of VLDL-bound triglycerides [26]. Additional alterations to hepatic clearance of VLDL remnants prolong their residence time and that of their constituent apo B (Figures 10 & 11).
Another section discusses the inflammation that characterizes visceral obesity. Both the inflammatory cells (macrophages) and the adipocytes themselves secrete many molecules, including pro-inflammatory cytokines (tumour necrosis factor-α, interleukin-6, resistin, etc.), that can affect distant tissues. Some of these cytokines reach the liver through the portal circulation and work with locally produced cytokines to alter hepatic lipid metabolism, exacerbating steatosis and VLDL triglyceride output into the bloodstream [27].
The discussion of fasting lipoprotein metabolism has so far focused on triglyceride production and secretion by the liver. However, insulin resistance is also caused by overproduction of triglyceride-rich lipoproteins by the intestine from endogenous fatty acids. This contributes to fasting hypertriglyceridemia [28], whose exact quantitative importance remains unknown.
In summary, the major causes of visceral obesity-related hypertriglyceridemia are as follows: fatty acid transfer from adipose tissue to the liver, adipose tissue insulin resistance, the production by adipose tissue of pro-inflammatory cytokines, liver insulin resistance and inflammation, increased liver VLDL synthesis and secretion, and reduced blood triglyceride clearance.
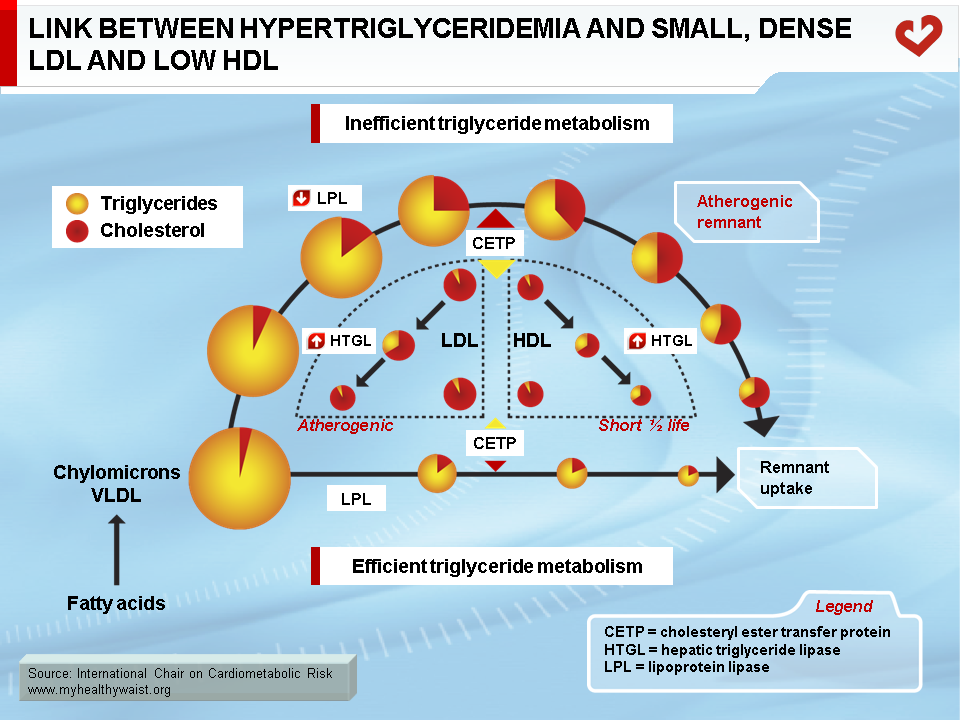
Metabolic Link Between Hypertriglyceridemia, Small, Dense LDL, and Low HDL
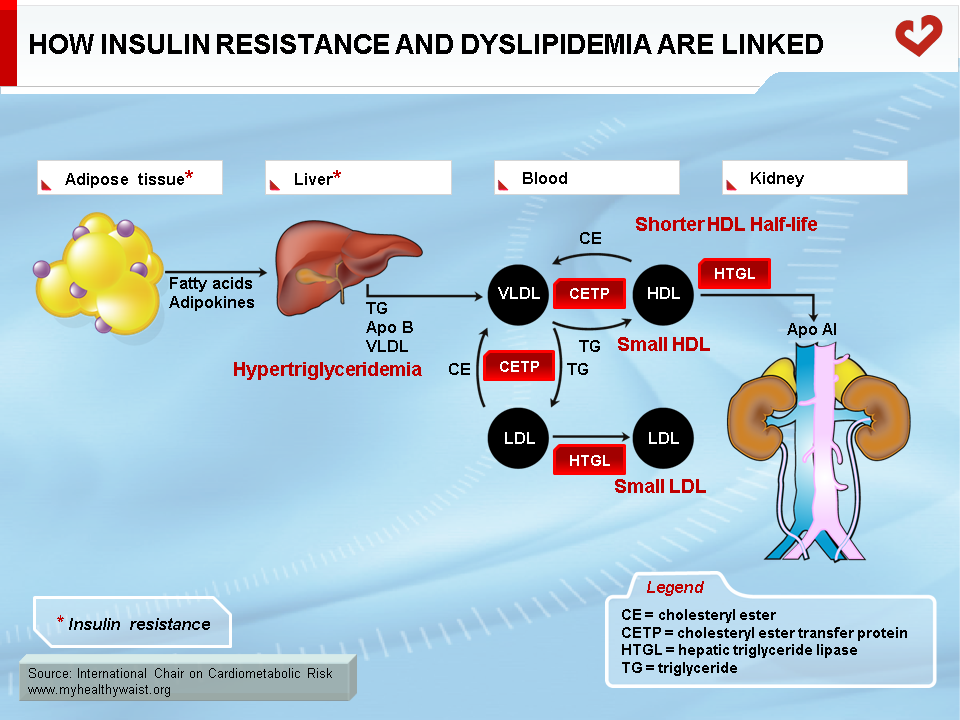
As discussed above, the obesity-related abnormalities due to VLDL, LDL, and HDL levels and composition are closely linked. The major cause of small LDL and lower HDL levels is the abnormal remodelling of these particles within the bloodstream by the enzymes CETP and HTGL [11,12]. First, because VLDL is more abundant, more LDL particles are generated through the VLDL remnant-LDL cascade. Second, because VLDL particles remain longer in the circulation, CETP has more time to mediate the exchange of lipids between lipoproteins: more triglycerides are transferred to cholesterol-rich LDL and HDL, and reciprocally more cholesterol to triglyceride-rich lipoproteins. Though normal LDL and HDL do not interact much with HTGL, triglyceride-enriched LDL and HDL become good substrates for HTGL, which hydrolyzes this excess load of triglycerides to produce smaller LDL and HDL particles. Small LDL particles bind less efficiently to LDL receptors, their normal route of clearance, which increases their residence time and number in the circulation. Insulin resistance worsens LDL clearance by removing insulin’s ability to stimulate expression of the LDL receptor.
As with triglyceride-enriched LDL, HTGL depletes HDL from excess triglycerides acquired by the enzyme CETP. This causes smaller HDL with lower than normal cholesterol relative to the protein components of the particles. For a variety of complex reasons, and contrary to small LDL, small HDL is more sensitive to degradation and is cleared faster from the circulation. This reduces HDL in the bloodstream and the total amount of cholesterol it carries (Figure 12) [29].
In summary, small LDL results from extensive LDL remodelling by CETP (triglyceride enrichment) and HTGL (triglyceride depletion and consequent decrease in size). HDL is remodelled in a similar way, making it more susceptible to degradation.
Major Characteristics of the Visceral Obesity-related Dyslipidemic Profile: Postprandial State
The above discussion focused on cholesterol- and triglyceride-carrying lipoproteins as they are produced and metabolized in the fasting state, i.e., from around 12 hours after cessation of caloric intake. However, eating three meals a day reduces the fasted state to only a few hours, and many people who snack late in the evening are in a non-stop postprandial state. It is therefore important to understand how excess visceral fat alters postprandial lipoprotein metabolism.
Visceral obesity and its frequent companion the metabolic syndrome are characterized by postprandial hypertriglyceridemia, defined as much higher than normal blood triglyceride concentrations following the ingestion of a standard mixed meal. The rate at which fat is absorbed by the intestine does not appear to play a major role. Instead, it is the blunted response to the postprandial rise in insulin that best explains postprandial hypertriglyceridemia. First, insulin resistance reduces the global availability of LPL in the capillary network and hence its ability to hydrolyze triglycerides. Second, it removes insulin’s usual ability to inhibit hepatic VLDL secretion, which causes the latter to continue instead of fall back after food ingestion. Unrestricted VLDL secretion by the liver and the appearance of gut-derived chylomicrons overwhelm the hydrolytic capacity of LPL, significantly delaying triglyceride clearance from the bloodstream. Third, insulin resistance hinders the anti-lipolytic action of insulin in adipose tissue so that lipolysis is not properly inhibited postprandially. High plasma fatty acids tend to hinder LPL activity, further delaying postprandial triglyceride clearance. Delayed triglyceride clearance in the postprandial state clearly plays a major role in the harmful remodelling of LDL and HDL discussed above [30].
In addition to hypertriglyceridemia, the postprandial state adds yet another dangerous player: the chylomicron remnant. Like VLDL remnants, these triglyceride-depleted, cholesterol-rich particles are generally short-lived because they are quickly cleared by the liver. However, their blood levels are higher in obese subjects. Remnants are highly atherogenic because they inhibit vasorelaxation, activate inflammation in endothelial cells, and are taken up by smooth muscle cells and macrophages to promote foam cell formation [31].
Thus, the major causes of postprandial hypertriglyceridemia are unopposed liver VLDL secretion, reduced intravascular LPL availability, and impairment of LPL hydrolytic action by high fatty acids and increased apo CIII content of triglyceride-rich lipoproteins.
Atherogenicity of the Dyslipidemia Associated with Visceral Obesity
Each component of the dyslipidemia described above contributes with many other factors to the development of atherosclerosis. It is likely that hypertriglyceridemia per se is an independent risk factor for atherosclerosis (i.e., risk unrelated to its impact on LDL and HDL remodelling), but this remains a matter of debate [32]. An established fact, however, is that remnants of triglyceride-rich lipoproteins, which are elevated in the hypertriglyceridemic state, are highly atherogenic, probably as much as small, dense LDL.
There are many reasons why small, dense LDL particles are atherogenic [33]. First, their longer residence time in the bloodstream due to high production and lower clearance rates encourages them to bind to the arterial wall. Second, changes in apo B on the surface of small LDL particles means they are more likely to interact with the surface of endothelial cells lining the arteries. This retention and the smaller size of LDL particles facilitate their entry into the vascular wall. Third, small LDL particles are very sensitive to chemical modification (oxidation, etc.) once inside the artery wall. Fourth, the receptors of resident macrophages recognize and take up modified LDL, which gradually turns these macrophages into foam cells. Foam cell formation is an early step in the development of atherosclerotic plaques.
In contrast, HDL is anti-atherogenic in a number of ways. HDL promotes cholesterol efflux from the arterial wall (as it does in all tissues) and its transport to the liver. HDL also prevents chemical modification of LDL within the artery wall, thereby reducing its uptake by macrophages. In addition, HDL hinders the processes that recruit macrophage precursors (monocytes) to the arterial wall, reducing the number of lipid-accumulating cells therein. Also, HDL particles carry molecules that possess anti-inflammatory, anti-thrombotic, and anti-oxidant properties, which reduces the inflammatory and oxidative burden in the artery wall as well as the formation of blood clots [34].
To summarize cholesterol movement under pro-atherogenic conditions, chylomicron remnants, VLDL remnants, and small LDL deliver cholesterol to the artery wall (pro-atherogenic), whereas HDL removes cholesterol from the artery wall (anti-atherogenic).
Dyslipidemia linked to visceral obesity is a major CVD risk factor and is one of the abnormalities upon which the definition of the metabolic syndrome is based. Hypertriglyceridemia is central to this dyslipidemic profile and has dire consequences on LDL and HDL composition and metabolism. In addition, visceral adipose tissue plays a major role in triglyceride metabolism. Many therapeutic options are available to treat dyslipidemia. However, targeting the fundamental cause of obesity-related dyslipidemia by reducing visceral fat undoubtedly remains an important therapeutic objective.
References
-
Genest J. Lipoprotein disorders and cardiovascular risk. J Inherit Metab Dis 2003; 26: 267-87.
PubMed ID: 12889666
-
Grundy SM. Cholesterol and Atherosclerosis, Diagnosis and Treatment. New York. Gower Medical Publishing. 1990.
PubMed ID:
-
Havel RJ and Kane JP. Structure and metabolism of plasma lipoproteins; the metabolic and molecular bases of inherited disease. New York. McGraw-Hill. 1995.
PubMed ID:
-
Ridker P, Genest J and Libby P. Plasma lipids and lipoproteins and cardiovascular diseases. Braunwald’s Heart Disease. New-York. Saunders. 2000.
PubMed ID:
-
Shelness GS and Sellers JA. Very-low-density lipoprotein assembly and secretion. Curr Opin Lipidol 2001; 12: 151-7.
PubMed ID: 11264986
-
Olivecrona T, Hultin M, Bergo M, et al. Lipoprotein lipase: regulation and role in lipoprotein metabolism. Proc Nutr Soc 1997; 56: 723-9.
PubMed ID: 9264122
-
Brown MS and Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232: 34-47.
PubMed ID: 3513311
-
Lamarche B and Paradis ME. Endothelial lipase and the metabolic syndrome. Curr Opin Lipidol 2007; 18: 298-303.
PubMed ID: 17495604
-
Schaefer EJ and Asztalos BF. Cholesteryl ester transfer protein inhibition, high-density lipoprotein metabolism and heart disease risk reduction. Curr Opin Lipidol 2006; 17: 394-8.
PubMed ID: 16832162
-
Hussain MM, Fatma S, Pan X, et al. Intestinal lipoprotein assembly. Curr Opin Lipidol 2005; 16: 281-5.
PubMed ID: 15891388
-
Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 2003; 46: 733-49.
PubMed ID: 12774165
-
Taskinen MR. Type 2 diabetes as a lipid disorder. Curr Mol Med 2005; 5: 297-308.
PubMed ID: 15892649
-
Yki-Jarvinen H. Fat in the liver and insulin resistance. Ann Med 2005; 37: 347-56.
PubMed ID: 16179270
-
Després JP, Lemieux I, Tchernof A, et al. [Fat distribution and metabolism]. Diabetes Metab 2001; 27: 209-14.
PubMed ID: 11452212
-
Després JP. Health consequences of visceral obesity. Ann Med 2001; 33: 534-41.
PubMed ID: 11730160
-
Després JP. Intra-abdominal obesity: an untreated risk factor for type 2 diabetes and cardiovascular disease. J Endocrinol Invest 2006; 29: 77-82.
PubMed ID: 16751711
-
Després JP. Is visceral obesity the cause of the metabolic syndrome? Ann Med 2006; 38: 52-63.
PubMed ID: 16448989
-
Després JP and Lemieux I. Abdominal obesity and metabolic syndrome. Nature 2006; 444: 881-7.
PubMed ID: 17167477
-
Grundy SM. Atherogenic dyslipidemia associated with metabolic syndrome and insulin resistance. Clin Cornerstone 2006; 8 Suppl 1: S21-7.
PubMed ID: 16903166
-
Lemieux S. Contribution of visceral obesity to the insulin resistance syndrome. Can J Appl Physiol 2001; 26: 273-90.
PubMed ID: 11441231
-
Pouliot MC, Després JP, Nadeau A, et al. Visceral obesity in men. Associations with glucose tolerance, plasma insulin, and lipoprotein levels. Diabetes 1992; 41: 826-34.
PubMed ID: 1612197
-
Després JP, Moorjani S, Lupien PJ, et al. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis 1990; 10: 497-511.
PubMed ID: 2196040
-
Pascot A, Lemieux I, Prud’homme D, et al. Reduced HDL particle size as an additional feature of the atherogenic dyslipidemia of abdominal obesity. J Lipid Res 2001; 42: 2007-14.
PubMed ID: 11734573
-
Lamarche B, Tchernof A, Mauriège P, et al. Fasting insulin and apolipoprotein B levels and low-density lipoprotein particle size as risk factors for ischemic heart disease. JAMA 1998; 279: 1955-61.
PubMed ID: 9643858
-
Frayn KN, Arner P and Yki-Jarvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem 2006; 42: 89-103.
PubMed ID: 17144882
-
Ruotolo G and Howard BV. Dyslipidemia of the metabolic syndrome. Curr Cardiol Rep 2002; 4: 494-500.
PubMed ID: 12379172
-
Mlinar B, Marc J, Janez A, et al. Molecular mechanisms of insulin resistance and associated diseases. Clin Chim Acta 2007; 375: 20-35.
PubMed ID: 16956601
-
Duez H, Lamarche B, Uffelman KD, et al. Hyperinsulinemia is associated with increased production rate of intestinal apolipoprotein B-48-containing lipoproteins in humans. Arterioscler Thromb Vasc Biol 2006; 26: 1357-63.
PubMed ID: 16614317
-
Lamarche B, Uffelman KD, Carpentier A, et al. Triglyceride enrichment of HDL enhances in vivo metabolic clearance of HDL apo A-I in healthy men. J Clin Invest 1999; 103: 1191-9.
PubMed ID: 10207171
-
Martins IJ and Redgrave TG. Obesity and post-prandial lipid metabolism. Feast or famine? J Nutr Biochem 2004; 15: 130-41.
PubMed ID: 15023394
-
Botham KM, Bravo E, Elliott J, et al. Direct interaction of dietary lipids carried in chylomicron remnants with cells of the artery wall: implications for atherosclerosis development. Curr Pharm Des 2005; 11: 3681-95.
PubMed ID: 16305504
-
Yuan G, Al-Shali KZ and Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ 2007; 176: 1113-20.
PubMed ID: 17420495
-
Rizzo M and Berneis K. Small, dense low-density-lipoproteins and the metabolic syndrome. Diabetes Metab Res Rev 2007; 23: 14-20.
PubMed ID: 17080469
-
Forrester JS and Shah PK. Emerging strategies for increasing high-density lipoprotein. Am J Cardiol 2006; 98: 1542-9.
PubMed ID: 17126667
 CLOSE
CLOSE
Genest J. Lipoprotein disorders and cardiovascular risk. J Inherit Metab Dis 2003; 26: 267-87.
PubMed ID: 12889666CLOSE
CLOSE
CLOSE
CLOSE
Shelness GS and Sellers JA. Very-low-density lipoprotein assembly and secretion. Curr Opin Lipidol 2001; 12: 151-7.
PubMed ID: 11264986CLOSE
Olivecrona T, Hultin M, Bergo M, et al. Lipoprotein lipase: regulation and role in lipoprotein metabolism. Proc Nutr Soc 1997; 56: 723-9.
PubMed ID: 9264122CLOSE
Brown MS and Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232: 34-47.
PubMed ID: 3513311CLOSE
Lamarche B and Paradis ME. Endothelial lipase and the metabolic syndrome. Curr Opin Lipidol 2007; 18: 298-303.
PubMed ID: 17495604CLOSE
Schaefer EJ and Asztalos BF. Cholesteryl ester transfer protein inhibition, high-density lipoprotein metabolism and heart disease risk reduction. Curr Opin Lipidol 2006; 17: 394-8.
PubMed ID: 16832162CLOSE
Hussain MM, Fatma S, Pan X, et al. Intestinal lipoprotein assembly. Curr Opin Lipidol 2005; 16: 281-5.
PubMed ID: 15891388CLOSE
Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 2003; 46: 733-49.
PubMed ID: 12774165CLOSE
Taskinen MR. Type 2 diabetes as a lipid disorder. Curr Mol Med 2005; 5: 297-308.
PubMed ID: 15892649CLOSE
Yki-Jarvinen H. Fat in the liver and insulin resistance. Ann Med 2005; 37: 347-56.
PubMed ID: 16179270CLOSE
Després JP, Lemieux I, Tchernof A, et al. [Fat distribution and metabolism]. Diabetes Metab 2001; 27: 209-14.
PubMed ID: 11452212CLOSE
Després JP. Health consequences of visceral obesity. Ann Med 2001; 33: 534-41.
PubMed ID: 11730160CLOSE
Després JP. Intra-abdominal obesity: an untreated risk factor for type 2 diabetes and cardiovascular disease. J Endocrinol Invest 2006; 29: 77-82.
PubMed ID: 16751711CLOSE
Després JP. Is visceral obesity the cause of the metabolic syndrome? Ann Med 2006; 38: 52-63.
PubMed ID: 16448989CLOSE
Després JP and Lemieux I. Abdominal obesity and metabolic syndrome. Nature 2006; 444: 881-7.
PubMed ID: 17167477CLOSE
Grundy SM. Atherogenic dyslipidemia associated with metabolic syndrome and insulin resistance. Clin Cornerstone 2006; 8 Suppl 1: S21-7.
PubMed ID: 16903166CLOSE
Lemieux S. Contribution of visceral obesity to the insulin resistance syndrome. Can J Appl Physiol 2001; 26: 273-90.
PubMed ID: 11441231CLOSE
Pouliot MC, Després JP, Nadeau A, et al. Visceral obesity in men. Associations with glucose tolerance, plasma insulin, and lipoprotein levels. Diabetes 1992; 41: 826-34.
PubMed ID: 1612197CLOSE
Després JP, Moorjani S, Lupien PJ, et al. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis 1990; 10: 497-511.
PubMed ID: 2196040CLOSE
Pascot A, Lemieux I, Prud’homme D, et al. Reduced HDL particle size as an additional feature of the atherogenic dyslipidemia of abdominal obesity. J Lipid Res 2001; 42: 2007-14.
PubMed ID: 11734573CLOSE
Lamarche B, Tchernof A, Mauriège P, et al. Fasting insulin and apolipoprotein B levels and low-density lipoprotein particle size as risk factors for ischemic heart disease. JAMA 1998; 279: 1955-61.
PubMed ID: 9643858CLOSE
Frayn KN, Arner P and Yki-Jarvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem 2006; 42: 89-103.
PubMed ID: 17144882CLOSE
Ruotolo G and Howard BV. Dyslipidemia of the metabolic syndrome. Curr Cardiol Rep 2002; 4: 494-500.
PubMed ID: 12379172CLOSE
Mlinar B, Marc J, Janez A, et al. Molecular mechanisms of insulin resistance and associated diseases. Clin Chim Acta 2007; 375: 20-35.
PubMed ID: 16956601CLOSE
Duez H, Lamarche B, Uffelman KD, et al. Hyperinsulinemia is associated with increased production rate of intestinal apolipoprotein B-48-containing lipoproteins in humans. Arterioscler Thromb Vasc Biol 2006; 26: 1357-63.
PubMed ID: 16614317CLOSE
Lamarche B, Uffelman KD, Carpentier A, et al. Triglyceride enrichment of HDL enhances in vivo metabolic clearance of HDL apo A-I in healthy men. J Clin Invest 1999; 103: 1191-9.
PubMed ID: 10207171CLOSE
Martins IJ and Redgrave TG. Obesity and post-prandial lipid metabolism. Feast or famine? J Nutr Biochem 2004; 15: 130-41.
PubMed ID: 15023394CLOSE
Botham KM, Bravo E, Elliott J, et al. Direct interaction of dietary lipids carried in chylomicron remnants with cells of the artery wall: implications for atherosclerosis development. Curr Pharm Des 2005; 11: 3681-95.
PubMed ID: 16305504CLOSE
Yuan G, Al-Shali KZ and Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ 2007; 176: 1113-20.
PubMed ID: 17420495CLOSE
Rizzo M and Berneis K. Small, dense low-density-lipoproteins and the metabolic syndrome. Diabetes Metab Res Rev 2007; 23: 14-20.
PubMed ID: 17080469CLOSE
Forrester JS and Shah PK. Emerging strategies for increasing high-density lipoprotein. Am J Cardiol 2006; 98: 1542-9.
PubMed ID: 17126667