Glucose/Insulin Homeostasis
Defining CMR - Visceral Adipose Tissue: the Culprit? Complications of Visceral ObesityKey Points
- Glucose is abundant in a wide range of foods. In the fasted state, the liver provides the bulk of glucose to the bloodstream through glycogenolysis and gluconeogenesis.
- Insulin optimizes glucose uptake by skeletal muscle, its major peripheral user. Insulin also reduces liver glucose production after a meal and reduces fatty acid release by adipose tissue. These three major functions are important for glucose homeostasis.
- Expansion of visceral fat causes adipocyte hypertrophy. This process triggers macrophages that, together with the enlarged adipocytes, locally secrete insulin-resistance-promoting molecules.
- Hypertrophied insulin-resistant visceral adipocytes release more fatty acids and proinflammatory adipokines into the bloodstream. The portal circulation carries these to the liver where they promote steatosis, insulin resistance, and local inflammation. The systemic circulation carries fatty acids and proinflammatory molecules to skeletal muscle where they promote lipid accumulation, insulin resistance, and local inflammation.
- Insulin resistance also affects the function of other systems and organs, including endothelial cells and cells of the vascular wall. This further increases CVD risk.
- Insulin resistance is believed to play a role in the development of many metabolic abnormalities that define the metabolic syndrome. It is also believed to be a strong link between visceral obesity and increased risk of type 2 diabetes and CVD.
- Targeting the fundamental cause of obesity-related insulin resistance by reducing visceral fat mass remains an important therapeutic objective.
Foreword
One of the most common metabolic complications of visceral obesity is insulin resistance, a condition in which insulin no longer functions effectively. It is generally accepted that insulin resistance is a major cause of many components of the metabolic syndrome, including dysregulation of glucose homeostasis, which can lead to type 2 diabetes. This is not surprising given that insulin is a hormone that plays a critical role in controlling carbohydrate, lipid, and protein metabolism and influences a host of other cellular functions in many organs.
This section explains the link between visceral obesity and insulin resistance in adipose and other insulin-responsive tissues, namely skeletal muscle and the liver, and how insulin resistance affects type 2 diabetes and cardiovascular disease (CVD) risk. A brief overview of how insulin regulates blood glucose is provided in order to place visceral obesity-related insulin resistance in proper context.
Glucose Metabolism
Sources of Glucose
Along with fatty acids, glucose is one of the body’s major energy sources. Virtually all body tissues use a mixture of glucose and fatty acids as substrates for ATP production, with some (brain, red blood cells) using glucose almost exclusively. Because the brain needs glucose continually, mammals have developed an exquisitely precise regulatory system to ensure that adequate amounts of glucose are always present within a narrow concentration range in the bloodstream.
In its free form or as part of more complex sugars, glucose is present in a wide range of foods. It is readily taken up from the gut and delivered to various tissues. Excess glucose is stored as glycogen, mainly in the liver and skeletal muscle. Some excess glucose can also be converted to fatty acids through a process called lipogenesis, mainly in the liver. The fatty acids are eventually delivered by lipoproteins to adipose tissue for long-term storage in the form of triglycerides.
Between meals, and especially at night, there is no incoming glucose from external sources, and the body must maintain blood glucose from endogenous sources. The major organ that provides glucose to the blood is the liver. A primary source of hepatic glucose is glycogen, a glucose polymer that is synthesized when plenty of glucose is available and provides a ready source of energy when necessary. In times of need, glycogen is hydrolyzed back into glucose molecules that are then exported to the bloodstream. A second important source of glucose comes from the synthesis of new glucose molecules from precursors such as glycerol (from hydrolysis of triglycerides in adipose tissue), lactate (from muscle anaerobic glycolysis), and some amino acids (from the breakdown of muscle proteins) through a process called gluconeogenesis. The latter occurs mainly in the liver, with the kidneys playing a small additional role. The rate of gluconeogenesis is driven mainly by the availability of precursors (which increase in the fasted state) and by the rate of fatty acid oxidation in the liver (also increased in the fasted state). If dietary nutrients are not incoming, the liver provides glucose to the bloodstream, ensuring a steady supply of fuel to the brain. Other tissues (e.g., muscle) also take up glucose in the fasted state but rely mainly on fatty acids (from hydrolysis of triglycerides in adipose tissue) as an energy substrate, thereby sparing glucose for use by the brain.
A detailed review of how glucose provides ATP is beyond the scope of this section and can be found in any biochemistry textbook (e.g., (1)). To summarize, once in the cell, glucose that is not stored as glycogen undergoes glycolysis, which yields a small amount of ATP. The glycolytic product pyruvate then migrates to the mitochondrion where it enters the Krebs cycle (also called the tricarboxylic, or TCA, cycle) to provide more ATP molecules.
Glucose Uptake by Tissues
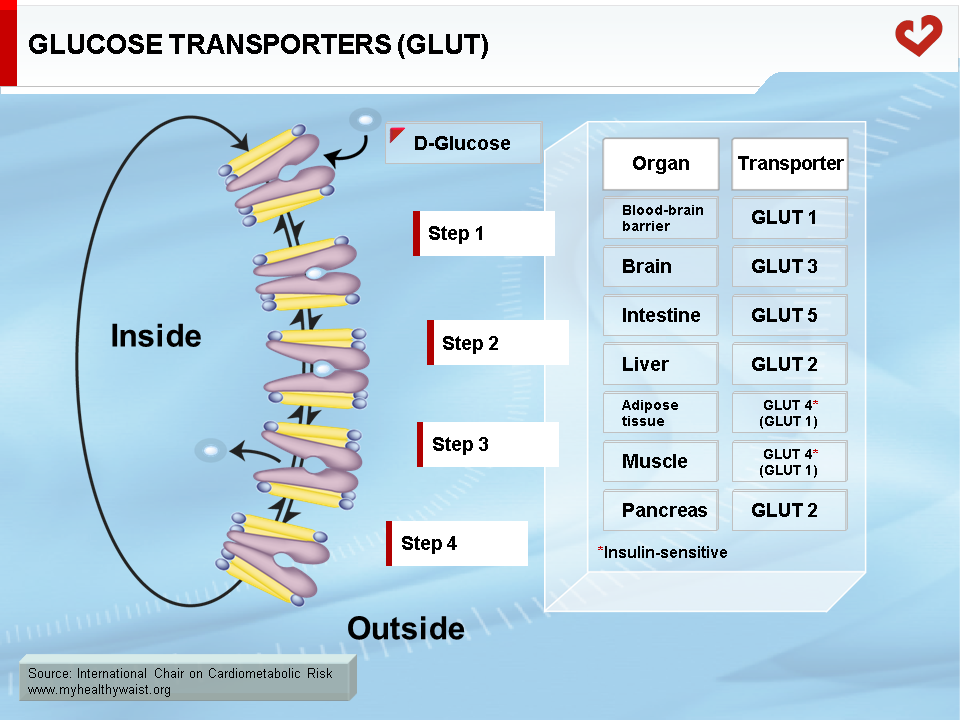
Glucose is carried freely in the blood and readily crosses endothelial cells that line blood capillaries to reach parenchymal cells. Although the glucose molecule is quite simple, it cannot cross the cell plasma membrane on its own and must be helped by membrane proteins called glucose transporters (GLUT). Several transporters are expressed in a tissue-specific manner (Figure 1). Most transporters function on their own, but GLUT4, the major isoform expressed in adipose tissue and skeletal muscle, requires insulin to function properly. This is very important because skeletal muscle is the major site (not considering the brain) of peripheral glucose disposal. Insulin-independent GLUT1 is also expressed in adipose tissue and muscle and mediates basal glucose uptake. GLUT4, however, is required to increase glucose uptake in times of plenty, after a meal for example. Proper glucose disposal is needed to maintain stable glycemia, and this requires efficient insulin action on GLUT4 function, mainly in skeletal muscle (2,3).
Role of Insulin in Glucose Homeostasis
Insulin, a hormone secreted by the beta cells of the pancreatic islets of Langerhans, plays a number of crucial roles in glucose homeostasis. Not only does it act directly on glucose uptake by insulin-sensitive tissues and on hepatic glucose production, it also acts indirectly by modulating adipose and liver lipid metabolism.
Control of Insulin Secretion
Insulin is secreted by pancreatic beta cells into the portal circulation (draining into the liver) at a low rate in the fasted state. The arrival of nutrients absorbed from the gut into the blood triggers a sharp rise in insulin output from the pancreas. The major insulin secretagogue is glucose. Incoming glucose is metabolized in the beta cell to ATP. This increases the ATP/ADP ratio, which closes ATP-sensitive K+ (K+ATP) channels, depolarizes (changes the voltage) the plasma membrane, opens voltage-dependent Ca2+ channels, and fosters Ca2+ triggering of insulin granule exocytosis (4). Other nutrients, including some fatty acids and amino acids, are also insulin secretagogues. Insulin secretion is further enhanced postprandially by the increase in gut hormones called incretins. Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are the most important, and their secretion is triggered by food ingestion. The brain also senses changes in blood glucose and modulates insulin secretion via parasympathetic and sympathetic nerve fibres to the pancreas (4).
The postprandial rise in insulin alters three major metabolic pathways related to glucose homeostasis: 1) insulin increases glucose uptake by insulin-sensitive tissues, mainly skeletal muscle, 2) insulin inhibits fatty acid release from adipose tissue, allowing glucose to become a major energy fuel, and 3) insulin decreases hepatic production of glucose, a sensible move given the arrival of dietary glucose. Following are the major mechanisms by which these important insulin-mediated metabolic changes take place.
Insulin and Tissue Glucose Uptake
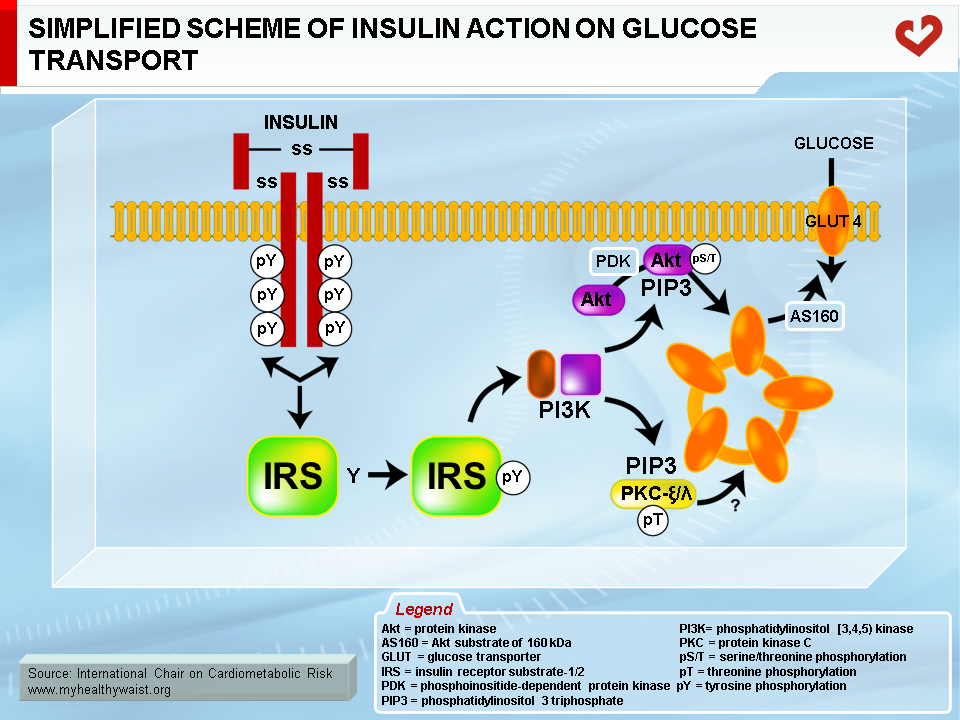
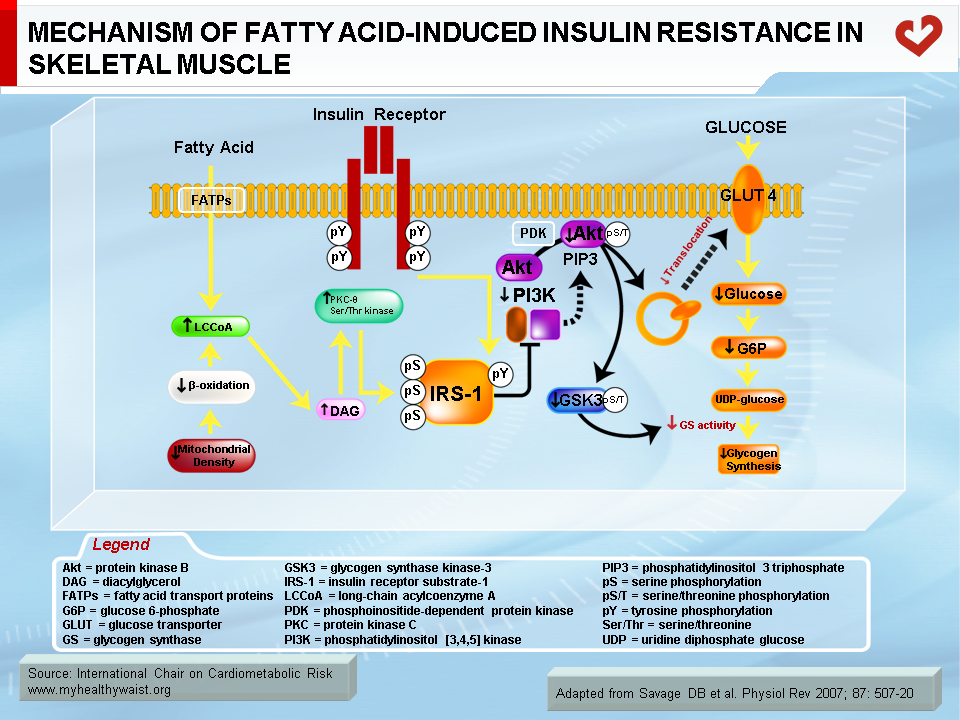
Skeletal muscle is the major organ where insulin-mediated glucose uptake occurs. Adipose tissue plays a smaller role. Insulin binds to skeletal muscle’s plasma membrane-bound receptor, which activates the phosphorylative function of the receptor. This triggers a cascade of phosphorylation on specific tyrosine residues of signalling proteins, downward from several insulin receptor substrates (IRS; IRS-1 is the major isoform in muscle), through phosphotidylinositol 3 kinase (PI3 kinase), to Akt (also known as Akt/protein kinase B, or Akt/PKB). This series of protein phosphorylations ultimately causes GLUT4 to migrate from intracellular vesicles to the plasma membrane, where it facilitates glucose transport into the cell (5) (Figure 2). The postprandial rise in insulin therefore favours the uptake of blood glucose by skeletal muscle (and to a lesser extent by adipose tissue) and gradually returns blood glucose to fasting levels.
Insulin and Adipose Tissue Lipolysis
In the fasted state, adipose tissue hydrolyzes some of its triglycerides into glycerol (a precursor of liver gluconeogenesis) and fatty acids (an important source of fuel for ATP production in various tissues). The postprandial rise in insulin acts on adipose tissue to inhibit lipolysis, which reduces the release of glycerol and fatty acids and encourages tissues such as skeletal muscle to use glucose as a fuel source. Normally, insulin strongly inhibits lipolysis by interfering with the action of hormone-sensitive lipase, a major lipolytic enzyme (6). Plasma levels of fatty acids then plummet.
Insulin and Liver Gluconeogenesis
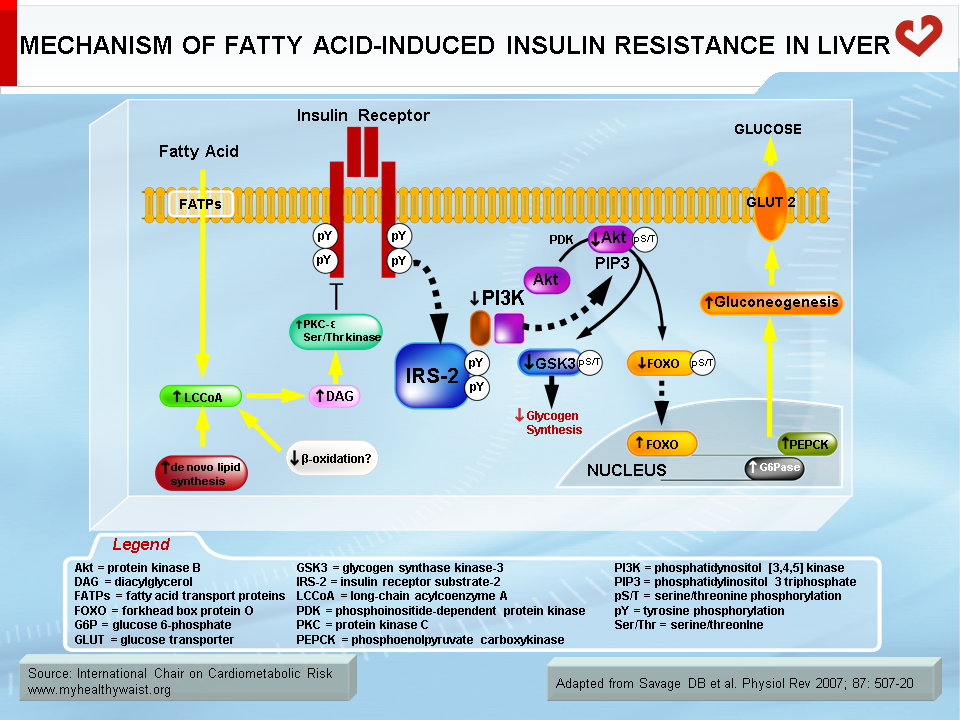
The increase in insulin that follows food intake reduces glucose production by the liver. This process is complex. Insulin negatively modulates the expression or activity of enzymes involved in gluconeogenesis (e.g., phosphoenolpyruvate carboxykinase, PEPCK) and glucose export (glucose-6-phosphatase, which produces exportable glucose) (7). Insulin also inhibits adipose tissue lipolysis, which reduces fatty acid delivery from visceral adipose tissue to the liver, lowers hepatic fatty acid oxidation rates, and thereby inhibits gluconeogenesis. How insulin acts directly (gluconeogenic enzyme expression/activity) and indirectly (adipose tissue fatty acid delivery) on gluconeogenesis is not yet fully understood.
Visceral Obesity and Insulin Resistance
One of the major conditions caused by excess visceral adipose tissue is insulin resistance, defined as the inability of insulin to perform many of its major metabolic functions. Most evidence points to a cause-and-effect relationship between excess visceral fat and insulin resistance, as well as between insulin resistance and the health complications associated with visceral obesity. It is therefore important to understand how visceral obesity leads to insulin resistance.
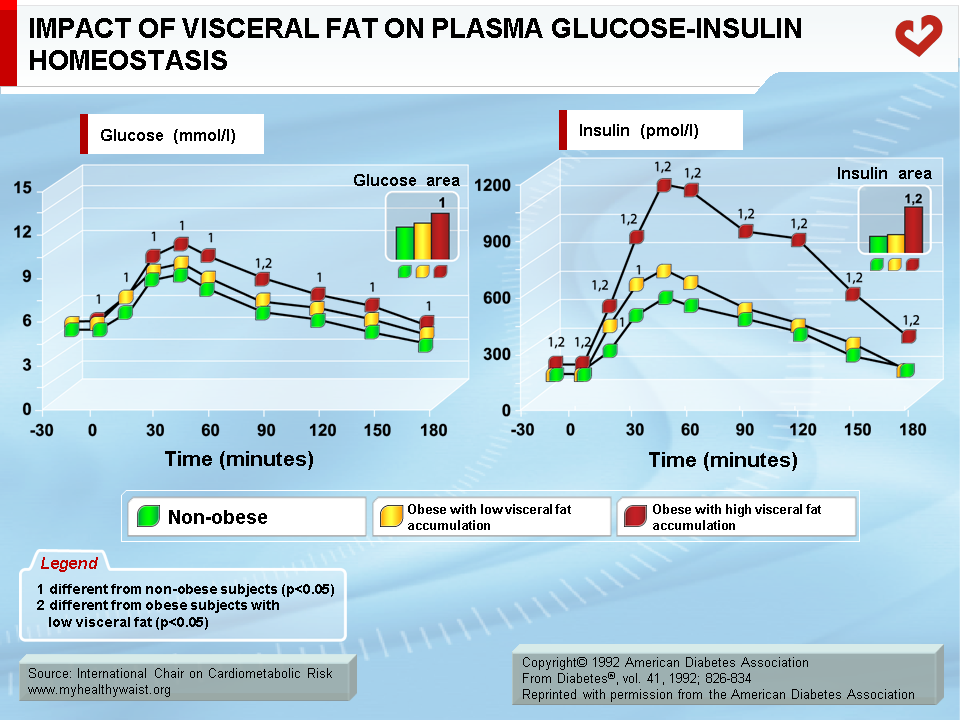
Key studies conducted in the early 1990s clearly established that it is the visceral component of excess fat, and not total fat, that is strongly associated with impaired insulin action (8,9). The glucose and insulin response to a glucose load was measured in men with identical amounts of total body fat and who were grouped according to whether they had low or high amounts of visceral fat (determined by computed tomography). Men with low visceral fat had a glucose and insulin response to the oral glucose challenge that was similar to that of a lean, control group of men. Men with high visceral fat had statistically greater glucose and insulin responses than men in the other two groups. These findings have since been confirmed in both men and women (Figure 3) (10,11).
Visceral Fat Cell Hypertrophy and the Immune Response
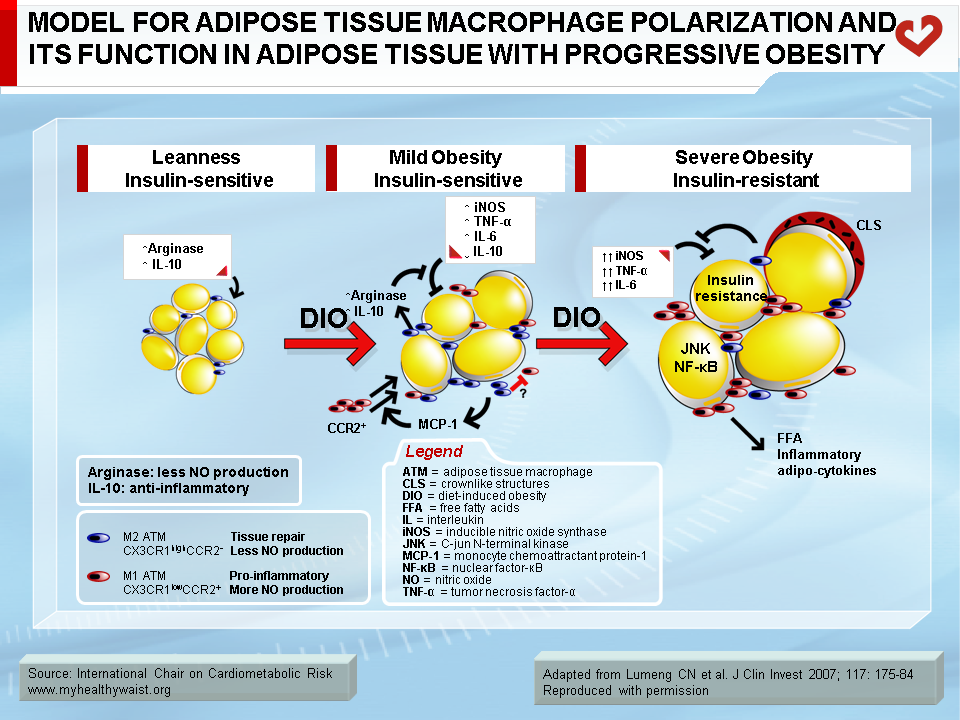
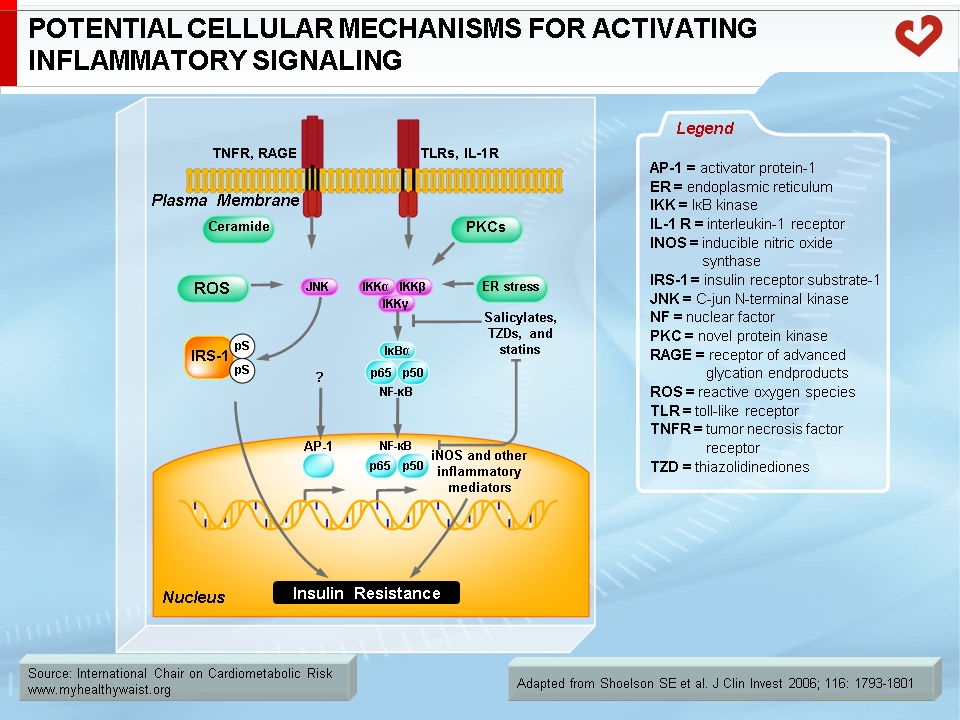
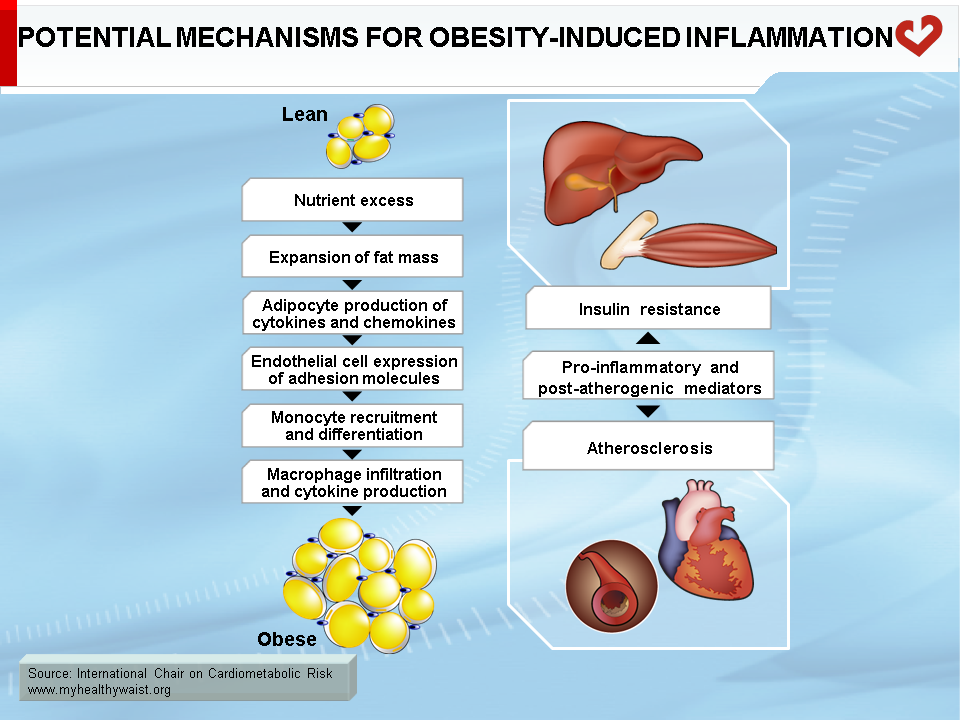
Under conditions that favour obesity, fat cells accumulate lipids to store excess energy and expand in size, a process known as hypertrophy. For reasons that are unclear, fat cells respond differently to hypertrophy depending on where they are located. Those in the subcutaneous compartments appear to be able to store more fat without much change in their overall metabolism. In visceral fat cells, however, a “stress response” occurs when a certain degree of hypertrophy is reached (12). This stress is ill-defined but may be related to excess energy substrates present within the adipocyte (12,13). The stress response includes the following: 1) activation of pathways (c-Jun NH2-terminal kinase (JNK) and NFkB, two master regulators of inflammation) that encourage the production of proinflammatory cytokines, 2) overproduction of reactive oxygen species (oxidative stress), and 3) production of signals for programmed cell death (apoptosis). The stress response has two major outcomes: 1) insulin resistance first develops in the adipocyte, which reduces glucose uptake and lessens inhibition of lipolysis and 2) the stressed adipocyte secretes molecules that chemically attract cells of the immune system, mainly monocytes (a type of white blood cell), that can become macrophages once within a tissue. Normally, resident macrophages protect tissues by taking up and eliminating potentially harmful cell debris and toxins. Macrophages in healthy tissues also secrete anti-inflammatory cytokines (e.g., interleukin-10 (IL-10)) and limit production of potentially dangerous molecules such as excess nitric oxide. In hypertrophic adipose tissue, however, alternate types of macrophages flourish. This encourages rather than inhibits inflammation and harms rather than protects the adipocyte, as discussed below (14).
One of several chemoattractant signals from hypertrophied adipocytes that has been given much attention lately is monocyte chemoattractant protein-1 (MCP-1, alternatively termed CCL-1) (15). Hypertrophied visceral adipocytes secrete this protein, which binds to a receptor (CCR-2) on the surface of monocytes in the bloodstream. The adipocyte stress signal (MCP-1) and its monocyte receptor interact to trigger a complex set of events that brings monocytes close to the adipocytes that initiated the signal, where they evolve into macrophages. MCP-1 is recognized as a powerful signal for initiating macrophage infiltration of “sick” adipose tissue. In rodent models, invalidation of the MCP-1 gene or that of its receptor CCR-2 partially protects against diet-induced insulin resistance (15,16).
Macrophage infiltration correlates with adipocyte size (17), and, as noted above, the events that trigger attraction of immune cells in the vicinity of expanding adipose cells appear to be relatively specific to visceral fat (18). The outcome of this process is adipose tissue inflammation, which is maintained by feed-forward cross-talk between the infiltrated immune cells and the adipocytes (19,20) (Figure 4). The inflammation associated with visceral obesity is discussed in more detail in another section.
Consequences of the Immune Response to Visceral Adipocyte Hypertrophy: Inflammation and Insulin Resistance
In adipose tissue
Interestingly, adipocytes and macrophages share many features, including the ability to secrete pro-inflammatory cytokines (tumour necrosis factor-α (TNF-α), IL-6, IL-8, and many others), MCP-1, and its receptor CCR-2, which act locally to influence adipocyte metabolism but are also released into the bloodstream and influence the metabolism of distant tissues. In the adipocyte, local inflammation leads to two major metabolic alterations: 1) increased fatty acid release into the bloodstream and 2) altered adipokine production that promotes inflammation and insulin resistance (TNF-α, IL-6, IL-8, resistin) while reducing insulin-sensitizing proteins (adiponectin) and anti-inflammatory (IL-10) adipokines (21). TNF-α (mainly released by macrophages but also by adipocytes) is thought to play a major role in causing these changes to adipocyte metabolism (6). TNF-α interferes with insulin signalling, decreasing glucose uptake, reducing insulin-mediated inhibition of lipolysis and fatty acid release into the circulation, and lowering the production of enzymes involved in lipid uptake and storage, which reduces the ability of adipocytes to clear lipids from the circulation. A number of factors appear to cause the changes in adipokine production, including TNF-α, MCP-1, and other cytokines derived from local macrophages. This process resembles a feed-forward cycle.
In the context of visceral obesity, excess fatty acids and proinflammatory cytokines, including adipokines from expanding adipose tissue, drive insulin resistance in non-adipose, insulin-responsive tissues. Although insulin resistance affects many metabolic pathways, the discussion that follows is limited to glucose metabolism.
In liver
Because lipolysis and inflammation are particularly severe in expanding visceral adipose depots and because these depots are partly drained by the portal vein, it is not surprising that the liver is a prime target for the problems caused by a dysfunctional visceral adipose tissue. Hepatic lipid accumulation (steatosis) often accompanies and parallels weight gain and visceral obesity. Steatosis triggers hepatic insulin resistance (as detailed below) and the expression of proinflammatory cytokines (e.g., TNF-α, IL-6, IL-1β) within liver cells, suggesting that lipid accumulation in the hepatocyte induces an inflammatory response similar to that seen with lipid accumulation in the adipocyte. The liver already contains a large amount of immune cells, such as Kuppfer cells (macrophages), that may amplify this inflammatory response within the hepatocyte. Proinflammatory cytokines delivered from visceral fat to the liver through the portal circulation may also spark liver inflammation. Lipid accumulation in the liver is not a benign condition and it can evolve into steatohepatitis or even cirrhosis in susceptible individuals. In addition, the inflamed liver secretes proinflammatory cytokines (TNF-α, IL-6, C-reactive protein) that are thought to help maintain the proinflammatory environment associated with visceral obesity (20).
In skeletal muscle
Excess fatty acids and proinflammatory cytokines from adipose tissue reach skeletal muscle through the systemic circulation, where they trigger insulin resistance. Although fatty acids released by visceral fat likely contribute to steatosis and insulin resistance in the liver, their contribution to systemic fatty acid levels and the amount of lipids that reach skeletal muscle is less clear. Some studies suggest that abdominal subcutaneous adipose tissue is the main contributor to systemic fatty acids (22). Visceral obesity is also characterized by hypertriglyceridemia, which may increase fatty acid delivery to muscle through local action of the lipoprotein lipase. With regard to proinflammatory cytokines, they may come from visceral fat depots and the liver (20) as well as from adipocytes found in the vicinity of skeletal muscle fibres.
Mechanisms of Fatty Acid-/Inflammation-induced Insulin Resistance
Normally, fatty acids taken up by skeletal muscle and the liver are either used as energy substrates to produce ATP through beta-oxidation or safely stored in the form of inert triglycerides. When fatty acids are in excess (a condition called ectopic fat accumulation), however, cells overproduce fatty acid metabolites such as fatty-acyl CoA, diacylglycerol, and ceramides. Some or all of these metabolites (depending on the tissue) trigger the activation of protein kinases that phosphorylate proteins involved in insulin signalling on their serine or threonine residues, rather than on tyrosine residues as normally occurs. This blocks instead of activates the signalling cascade (23) (Figures 5 and 6). An obvious source of excess fatty acids is enlarged adipose tissue. However, defects in local mitochondrial function that impair fatty acid oxidation have also been held out as causes of intracellular accumulation of insulin resistance-promoting fatty acid derivatives (23).
Together with fatty acid metabolites, proinflammatory adipokines that are overproduced by expanding visceral fat and other possible sources (see above) also play a role in the development of insulin resistance. The mechanisms that link proinflammatory cytokines and insulin signalling are complex and not fully understood. Some inflammatory signals such as TNF-α are thought to directly alter the phosphorylation of proteins of the insulin-signalling cascade (e.g., serine instead of tyrosine phosphorylation of IRS-1) by activating protein kinases such as JNK. Others initiate the proinflammatory signalling cascades governed by the JNK and the IKK-beta–NFκB pathways, leading to further expression of inflammatory proteins and local insulin resistance (19,20) (Figure 7).
From Insulin Resistance to Type 2 Diabetes
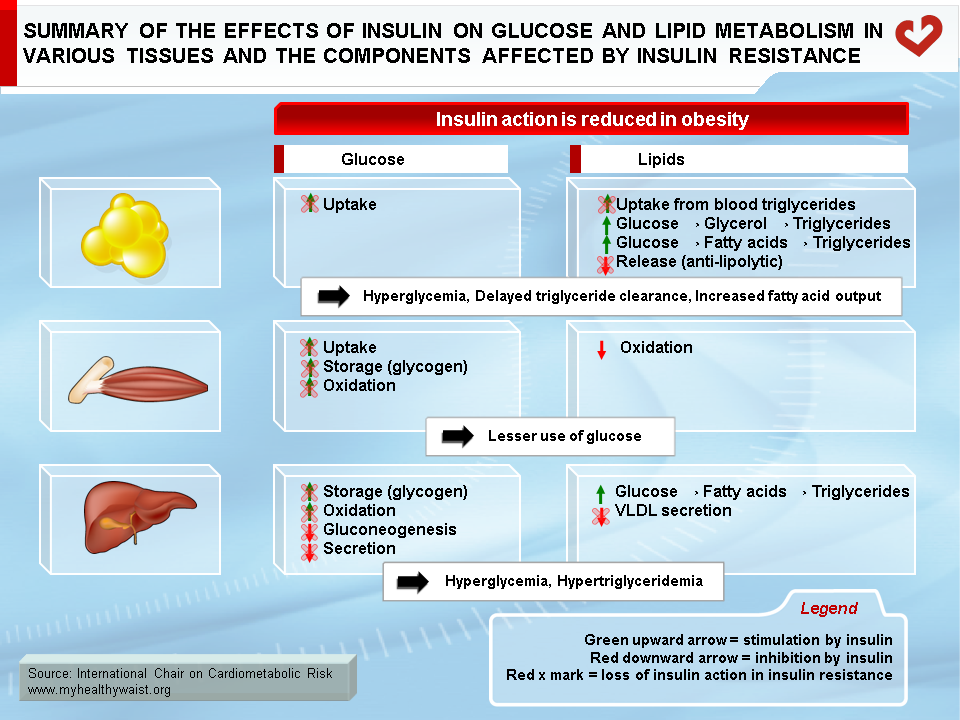
In insulin-resistant skeletal muscle, insulin’s positive effect on GLUT4 translocation and glucose uptake is reduced. In insulin-resistant subjects, glucose uptake by skeletal muscle, a major site of its clearance, does not increase after a meal, which leaves more glucose in the bloodstream. In the insulin-resistant liver, insulin is unable to reduce de novo glucose production (gluconeogenesis) and glucose export to the circulation, which hinders the postprandial reduction in hepatic glucose output and further increases circulating glucose. The final outcome is that the body is less able to tailor glucose production and clearance to the prevailing metabolic conditions.
The pancreas has the remarkable ability to adjust insulin secretion in an attempt to overcome insulin resistance. If insulin becomes less efficient at handling glucose, pancreatic beta cells become more active and secrete more insulin to compensate for insulin resistance. Several factors contribute to islet compensation, including increased glucose oxidation within the beta cell, increased fatty acid signalling (because of enhanced release by visceral adipose tissue), sensitivity to intestinal incretins, and increased parasympathetic nervous system activity in islets (24). Compensatory hyperinsulinemia is able, at least for a while, to keep blood glucose levels within the normal range in the fasted and postprandial states. This is because the extra insulin compensates for its reduced ability to stimulate muscle (and adipose tissue) glucose uptake and reduce hepatic glucose production. At this stage, the glycemic profile therefore appears normal and does not provide information on an individual’s metabolic status. Only assessing insulin levels in the fasted state and in response to glucose or mixed-food intake can indicate the existence of insulin resistance, which will appear as hyperinsulinemia in the postprandial state or in both fasting and postprandial states, depending on the severity of insulin resistance.
In individuals with robust pancreatic beta cells, insulin resistance may become more severe, with the pancreas remaining able to produce and secrete ever more insulin to compensate for its deteriorating peripheral action. These individuals do not present with abnormal glycemia. In other individuals, however, pancreatic beta cells do not continue compensating with extra insulin over time. A detailed review of the causes and mechanisms of beta cell deterioration is beyond the scope of this section. Several metabolic triggers of insulin resistance mentioned above (e.g., excess fatty acids, inflammation) likely also play a role in beta cell deterioration (25). Though not fully understood, genetic predisposition and environmental factors determine beta cell fragility upon exposure to stress.
The mildest form of glucose dysregulation caused by insulin resistance is impaired glucose tolerance (IGT). The condition manifests as abnormal handling of a glucose load (whether pure or as part of a regular meal). Subjects with IGT have normal fasting glucose (thanks to compensatory hyperinsulinemia), but an exaggerated increase in blood glucose in response to glucose ingestion. The increased glucose indicates that the pancreas is unable to produce enough insulin to fully handle the glucose load within a normal timeframe. Obesity is frequently associated with IGT, and studies have clearly established that it is excess visceral fat rather than excess overall fat that is associated with the condition (8-11).
As insulin resistance worsens and/or pancreatic beta cell deterioration progresses, not enough insulin may be secreted to maintain normal glucose levels, even in the fasted state. This condition is impaired fasting glucose (IFG). Here glycemia measured after an overnight fast is higher than normal but still below the threshold for type 2 diabetes.
In some individuals, whole-body insulin resistance and beta cell damage become so severe that the residual insulin that may still be produced has little or no effect on glucose handling. Type 2 diabetes, defined as fasting glycemia higher than the consensus level, soon sets in. Various healthcare organizations (e.g. (26)) have published glycemic thresholds and other information regarding diagnosis and definition of conditions related to glucose and insulin homeostasis.
Despite the fact that insulin resistance is a risk factor for type 2 diabetes, most insulin-resistant individuals will never develop type 2 diabetes. The hyperinsulinemia resulting from compensatory hypersecretion by the pancreas keeps glycemia within the normal range. However, epidemiological data shows that, even if blood sugar levels are normal, the CVD risk resulting from the metabolic abnormalities associated with the metabolic syndrome (of which insulin resistance is a central component) increases significantly.
Consequences of Abnormal Glucose and Insulin Homeostasis on CVD Risk
Hyperglycemia
Hyperglycermia (be it postprandial in IGT or daylong in IFG and type 2 diabetes) has several harmful effects in and of itself. As with fatty acids, excess glucose contributes to insulin resistance and is toxic to beta cells (glucotoxicity) (27,28). Hyperglycemia leads to the formation of advanced glycated end-products (AGE), which are caused by the nonenzymatic reaction of glucose with proteins in the bloodstream. High blood glucose and AGE levels are known to increase atherogenicity (29,30). In addition, prolonged, high levels of glucose can cause retinopathy, neuropathy, nephropathy, and CVD to a lesser extent (31-33). In clinical practice, long-term exposure to hyperglycemia is evaluated by measuring glycosylated hemoglobin (HbA1C). Typically, diabetic patients have plasma HbA1C levels over 6%. The UK Prospective Diabetes Study has clearly shown that reducing hyperglycemia decreases the incidence of diabetes-related complications. Each 1% decrease in blood HbA1C reduces the risk of diabetes-related complications by about 21%, diabetes-related mortality by 21%, myocardial infarction by 14%, and microvascular complications by 37% (31).
Hyperinsulinemia
The chronic hyperinsulinemia needed to compensate for insulin resistance also has harmful health consequences. Constant exposure to high insulin may desensitize its signalling pathway (i.e., insulin resistance) in target tissues (19, 20), creating a vicious cycle between the hormone and its function. High insulin has also been shown to have a direct, negative impact on endothelial function and vascular biology, which raises overall cardiovascular health risk (34). Lowering insulinemia per se is not a therapeutic option, as the tradeoff is diabetes. Instead, it is insulin resistance that must be the priority target when treating insulin homeostasis problems.
Insulin resistance
Beyond blood levels of glucose and insulin, it is insulin resistance that appears to bear most of the blame for the heightened type 2 diabetes and CVD risk that comes with visceral obesity. Insulin resistance is at the very core of many components of the metabolic syndrome, including major CVD risk factors such as dyslipidemia, hypertension, and type 2 diabetes. In addition, insulin resistance affects not only glucose and lipid metabolism (summarized in Figure 8), but also a host of other cellular functions that extend far beyond “metabolic” tissues. These factors working in unison increase CVD risk. As described above and depicted in Figure 9, insulin resistance is due in large part to visceral fat expansion, which triggers a local inflammatory reaction and increased fatty acids that spread to other organs and systems.
Lifestyle and pharmacological treatments exist to control glycemia and insulin action. However, targeting the fundamental cause of obesity-related insulin resistance by reducing visceral fat undoubtedly remains a key therapeutic objective.
References
-
Murray R, Mayes PA and Rodwell VW. Harper’s Biochemistry. McGraw-Hill Companies. 2003.
PubMed ID:
-
Huang S and Czech MP. The GLUT4 glucose transporter. Cell Metab 2007; 5: 237-52.
PubMed ID: 17403369
-
Thirone AC, Huang C and Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab 2006; 17: 72-8.
PubMed ID: 16458527
-
Prentki M. New insights into pancreatic beta-cell metabolic signaling in insulin secretion. Eur J Endocrinol 1996; 134: 272-86.
PubMed ID: 8616523
-
Chang L, Chiang SH and Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med 2004; 10: 65-71.
PubMed ID: 16307172
-
Arner P. The adipocyte in insulin resistance: key molecules and the impact of the thiazolidinediones. Trends Endocrinol Metab 2003; 14: 137-45.
PubMed ID: 12670740
-
Quinn PG and Yeagley D. Insulin regulation of PEPCK gene expression: a model for rapid and reversible modulation. Curr Drug Targets Immune Endocr Metabol Disord 2005; 5: 423-37.
PubMed ID: 16375695
-
Després JP, Moorjani S, Lupien PJ, et al. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis 1990; 10: 497-511.
PubMed ID: 2196040
-
Pouliot MC, Després JP, Nadeau A, et al. Visceral obesity in men. Associations with glucose tolerance, plasma insulin, and lipoprotein levels. Diabetes 1992; 41: 826-34.
PubMed ID: 1612197
-
Ross R, Aru J, Freeman J, et al. Abdominal adiposity and insulin resistance in obese men. Am J Physiol Endocrinol Metab 2002; 282: E657-63.
PubMed ID: 11832370
-
Ross R, Freeman J, Hudson R, et al. Abdominal obesity, muscle composition, and insulin resistance in premenopausal women. J Clin Endocrinol Metab 2002; 87: 5044-51.
PubMed ID: 12414870
-
Gregor MG and Hotamisligil GS. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J Lipid Res 2007; 48: 1905-14.
PubMed ID: 17699733
-
Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006; 444: 860-7.
PubMed ID: 17167474
-
Lumeng CN, Bodzin JL and Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007; 117: 175-84.
PubMed ID: 17200717
-
Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006; 116: 1494-505.
PubMed ID: 16691291
-
Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 2006; 116: 115-24.
PubMed ID: 16341265
-
Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796-808.
PubMed ID: 14679176
-
Yu R, Kim CS, Kwon BS, et al. Mesenteric adipose tissue-derived monocyte chemoattractant protein-1 plays a crucial role in adipose tissue macrophage migration and activation in obese mice. Obesity (Silver Spring) 2006; 14: 1353-62.
PubMed ID: 16988077
-
Shoelson SE, Herrero L and Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007; 132: 2169-80.
PubMed ID: 17498510
-
Shoelson SE, Lee J and Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006; 116: 1793-801.
PubMed ID: 16823477
-
Skurk T, Alberti-Huber C, Herder C, et al. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab 2007; 92: 1023-33.
PubMed ID: 17164304
-
Koutsari C and Jensen MD. Thematic review series: patient-oriented research. Free fatty acid metabolism in human obesity. J Lipid Res 2006; 47: 1643-50.
PubMed ID: 16685078
-
Savage DB, Petersen KF and Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev 2007; 87: 507-20.
PubMed ID: 17429039
-
Prentki M and Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006; 116: 1802-12.
PubMed ID: 16823478
-
Kahn SE, Hull RL and Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006; 444: 840-6.
PubMed ID: 17167471
-
American Diabetes Association. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2020. Diabetes Care 2020; 43 (Suppl. 1): S14-S31.
PubMed ID: 31862745
-
Rossetti L, Giaccari A and DeFronzo RA. Glucose toxicity. Diabetes Care 1990; 13: 610-30.
PubMed ID: 2192847
-
Stumvoll M, Goldstein BJ and van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005; 365: 1333-46.
PubMed ID: 15823385
-
Yan SF, Yan SD, Herold K, et al. Receptor for advanced glycation end products and the cardiovascular complications of diabetes and beyond: lessons from AGEing. Endocrinol Metab Clin North Am 2006; 35: 511-24, viii.
PubMed ID: 16959583
-
Bansilal S, Farkouh ME and Fuster V. Role of insulin resistance and hyperglycemia in the development of atherosclerosis. Am J Cardiol 2007; 99: 6B-14B.
PubMed ID: 17307054
-
Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000; 321: 405-12.
PubMed ID: 10938048
-
Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352: 854-65.
PubMed ID: 9742977
-
Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352: 837-53.
PubMed ID: 9742976
-
Nitenberg A, Cosson E and Pham I. Postprandial endothelial dysfunction: role of glucose, lipids and insulin. Diabetes Metab 2006; 32 Spec No2: 2S28-33.
PubMed ID: 17375404
